Page: /








4) Sleep Disturbances and CFS as sleep disorder continued
overview, see https://bra.in/4p6d3Q
![]()
5) Overview
CFS and Neurology An overview of recent research https://www.prohealth.com/library/cfs-and-neurology-an-overview-of-recent-research-19550
Many observations suggest that CFS could derive from residual damage to the reticular activating system (RAS) of the upper brain stem and/or to its cortical projections. It should be pointed out that although the larger right greater than left asymmetry in regional cerebral blood flow is found at the parietotempotal level in CFS patients as compared to healthy controls, no significant correlations are found between frontal tracer uptake and right-left parietotemporal asymmetry, on the one hand, and clinically relevant CFS dimensions on the other. Damage to the RAS could be produced by a previous viral infection, leaving functional defects unaccompanied by any gross histological changes.
In this respect, fluorine-deoxyglucose positron emission tomography showed specific metabolism abnormalities in CFS patients (hypometabolism in right mediofrontal cortex and brainstem) as compared with both healthy controls and depressed patients. The most relevant abnormality is brain stem hypometabolism, which has been also reported in single-photon emission computed tomography studies and seems to be a marker for the in vivo diagnosis of CFS

click
Fatigue diagrams part 2
please wait for document to load, use sidebar to scroll down page and 'Previous' and 'Next' buttons to advance pages
click here to read the entire document as a continuous file (or if document takes too long to load)
6) CFS similarities analogy to Hibernation state
Decreases in Sphingolipid, glycosphingolipid, phospholipid, purine, microbiome aromatic amino acid and branch chain amino acid metabolites, as well as in flavine adenine nucleotide (FAD) and lathosterol
7) Apnea
Untreated sleep apnea causes daytime tiredness, even with a full night’s sleep
I have mild sleep apnea and find the CPAP machine difficult to sleep with. I will try again. In the meantime, perhaps snore surgery reduces apnea? I've been using a CPAP machine for a couple months now and still haven't gotten used to it (I find it disruptive to my sleep). Seeing as it is logical to use (and my snoring bothers my fiance') I will continue to use it until I get used to it or until I opt for surgery. I found that keeping the air pressure to a low setting helps you to acclimate to the device (otherwise subconsciously I yank it off in a half sleep state
7b) Apnea Surgery
Tonsillectomy for Sleep Apnea as First-Line Treatment in Adults https://sleep-doctor.com/blog/tonsillectomy-for-sleep-apnea-as-first-line-treatment-in-adults
For adults with obstructive sleep apnea, the standard treatment is positive airway pressure therapy (such as CPAP, BPAP, or APAP). Surgery is reserved for patients who are unable to tolerate or benefit from positive airway pressure therapy. For children, adenoidectomy and/or tonsillectomy for sleep apnea is the standard treatment. Positive airway pressure therapy is not an ideal treatment for most children. This is due to concerns over effects on facial growth and difficulty that children may have with tolerating it through the night. It is reassuring that surgical outcomes in children–while by no means perfect–are relatively good, especially when the tonsils or adenoids are enlarged and when the child is not considered substantially overweight.
What about tonsillectomy for sleep apnea as first-line treatment in adults?
Just like in children, adults with enlarged tonsils also do better after sleep apnea surgery that includes tonsillectomy. One reason seems to be that the physical removal of the enlarged tonsils immediately opens up space for breathing and improves the sleep apnea. Many have wondered whether adults with sleep apnea and markedly enlarged tonsils should be treated with surgery that includes tonsillectomy.
The December 2016 issue of the medical journal The Laryngoscope included an interesting study examining this question. Twenty-nine adults with markedly enlarged tonsils (size 3+ or 4+ on the Friedman scale), obstructive sleep apnea, and no substantial obesity (body mass index below 32 kg/meters squared) underwent tonsillectomy alone. One patient was lost to follow up, but the rest of the patients had sleep studies before and then 6 months after surgery. Impressively, the average apnea-hypopnea index decreased from 40 to 7 events per hour after undergoing tonsillectomy for sleep apnea, with only 2 patients having anything worse than mild sleep apnea. There were also substantial improvement in the score on the Epworth Sleepiness Scale score that measures daytime sleepiness (mean score decreased from 11 to 6
Are there other studies of tonsillectomy for sleep apnea?
This study followed previous smaller studies showing substantial improvement or resolution in sleep apnea after tonsillectomy alone and that tonsil size and body mass index were associated with outcomes after tonsillectomy alone and that tonsillectomy could reduce the required CPAP pressure in those who did not have resolution of their sleep apnea. This was supported by a larger study of 202 adults published in 2015. This study showed a 95% chance of surgical success after tonsillectomy for sleep apnea, with a decrease in the average apnea-hypopnea index from 18 to 3 events per hour.
So why isn’t tonsillectomy for sleep apnea a first-line treatment in adults?
There are likely many reasons. First, not all patients have tonsils that are markedly enlarged. I would estimate that this about 5-10% of all adults with sleep apnea would be ideal candidates for tonsillectomy as a first-line treatment. This figure seems relatively small, but it still is quite a few patients who could have their tonsils removed because sleep apnea is so common. Second, most of these studies are relatively small. It would be important to repeat the studies in larger groups, just to confirm the findings. Third, the studies are not what are called randomized trials. Randomized trials could include patients with sleep apnea and markedly enlarged tonsils, either performing tonsillectomy or observing them without treatment for a period of time (6 months, for example). Unfortunately, it turns out that making people wait for surgery just to be part of a research study is incredibly difficult. Patients will prefer not to be involved in these studies if they are interested in having surgery (or any treatment). Finally, there are perceptions about surgery for sleep apnea that we have to overcome. I have written before that most surgeons, other physicians, and the public think that there is only one surgery for sleep apnea. That is just not the case.
What would I recommend?
We are in the midst of a major change in rethinking sleep apnea surgery–for all parties involved. The goal is developing a tailored approach to sleep apnea treatment with an approach that is often called personalized medicine. I see many young adults with markedly enlarged tonsils who are struggling with positive airway pressure therapy, including many with mild sleep apnea who are not overweight. For these patients, I think it is very reasonable to think about surgery as a first-line option instead of being on positive airway pressure for the rest of their life. These patients have a greater than 90% chance of clearing up their sleep apnea with tonsillectomy alone. Not every one of them will want to have surgery, but this should be part of the discussion because the results will be so good, based on everything we know about sleep apnea surgery outcomes.
As a sleep surgeon, I see many patients who want surgery because they simply do not like positive airway pressure therapy, even though they are doing well with it. In fact, I actually discourage many of these patients from surgery. My approach is always the same: if you are doing well with positive airway pressure therapy, keep using it. The one caveat are those patients who have a very high chance of resolution of their sleep apnea with a straightforward procedure like tonsillectomy
Treatment considerations for excessive daytime sleepiness (EDS) in OSA https://edsandosa.com
- While continuous positive airway pressure (CPAP) is considered the gold standard of treatment for obstructive sleep apnea (OSA). EDS may persist despite optimal CPAP use
- Sleep apnea treatments like CPAP have been proven to decrease symptoms such as apneas, hypopneas, and snoring, and improve sleep structure in patients with OSA.
- However, CPAP and CPAP alternatives may not address the brain alterations and subsequent neurologic dysfunction that OSA can leave behind
Pharmacotherapy should be considered for patients who have unresolved EDS despite optimal CPAP compliance'°
7c) CPAP and surgery alternatives
CPAP alternatives include bilevel positive airway pressure (BPAP). automatic (or autotitrating) positive airway pressure (APAP), oral appliances, and upper airway stimulation devices.'
7e) Neurological Effects of Apnea
Emerging science can help explain the link between excessive daytime sleepiness (EDS) and OSA
- EDS is one of the most common symptoms of obstructive sleep apnea (OSA)
- Animal and human studies suggest that the recurring cycle of intermittent hypoxia and sleep fragmentation associated with OSA may result in changes to the brain
- The subsequent disruption in neurologic function + may manifest as excessive sleepiness during the day
- While brain alterations have been linked to sleepiness, persistent EDS may be due to other factors such as chronic sleep loss and coc disorders1"
In an animal model of severe sleep apnea, chronic intermittent hypoxia led to significant neuronal injury'
Long-term hypoxia significantly increased oxidative injury to wake-promoting dopaminergic and noradrenergic neurons compared with the control group (Pc-0.01)2*
The loss of medial dendrites represents irreversible and functionally significant injury to wake-promoting regions of the brain'
At 6 months, a 40% JOSS of select wake-promoting dopaminergic and noradrenergic neurons was associated with irreversible wake impairments'
In a separate animal model of OSA, chronic sleep fragmentation led to a significant loss of wake-promoting neurone
Exposure to sleep disruptions over 14 weeks caused a significant reduction of wake-promoting noradrenergic neurons compared with the control group (P<0.001), even after 4 weeks of recovery'
Excessive daytime sleepiness (EDS) in OSA is associated with significant changes to the brain
Obstructive sleep apnea (OSA) is associated with reduced gray matter concentration"
Imaging studies in patients with severe OSA showed reduced gray matter concentration in certain brain regions compared with healthy volunteers, including the frontal cortex, anterior cingulate cortex, and thalamus.''
Some of these regions are involved in wakefulness and neurocognitive performance in' • Problem-solving • Planning • Decision-making • Attention/concentration
EDS in OSA is associated with significant white matter structural alterations despite the optimal use of CPAP'
Widespread white matter changes were observed in patients with OSA-associated EDS despite optimal CPAP adherence hours for 1 month) vs nonsleepy patients'
These changes ideate potential myelin damage and compromised neuronal connectivity'
This may help explain why EDS can persist in patients with OSA, even with optimal CPAP use?
Excessive daytime sleepiness (EDS) in OSA is more common than you may think
- Animal and human studies indicate a link between obstructive sleep apnea (OSA) and changes to the brain—and these changes have been associated with a disruption in neurologic function'
- While CPAP is needed to address the airway issue in patients with OSA, it may not address all aspects of compromised neuronal activity, and EDS may persist"
In a study of patients with OSA,
1 IN 3 WHO USED THEIR CPAP 3 HOURS A NIGHT STILL REPORTED FEELING SLEEPY DURING THE DAY"
`A study of 174 patients with moderate to severe OSA using continuous positive airway pressure (CPAP). Daytime sleepiness was assessed before and after 3 months of CPAP therapy using the Epworth Sleepiness Scale.' 'Includes average CPAP use of 2 or fewer hours per night, up to 7 or more hours per night.'
8) Hypothalamus
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome as Disturbed Homeostasis due to Focal Inflammation in the Hypothalamus (click to open)http://jpet.aspetjournals.org/content/jpet/early/2018/08/03/jpet.118.250845.full.pdf Chronic fatigue syndrome and disturbed homeostasis AbbreviationsADP=adenosine diphosphateAMPK=5’ adenosine monophosphateactivated protein kinaseApoE= Apolipoprotein EAT=anaerobic thresholdATP=adenosine5’\triphosphateANS= autonomic nervous system BMI=bodymass index βFGF=βfibroblast growth factor CGRP= calcitoningene related proteinCNS=central nervous systemCRH= corticotropinreleasing hormoneCSF= cerebrospinal fluidCVD= cardiovascular diseaseFAD=flavine adenine nucleotideFMS= fibromyalgia syndromeGWI= Gulf War IllnessHDL=highdensity lipid (cholesterol)HPA=hypothalamicpituitaryadrenal axisIBS=irritable bowel syndromeIFNγ= interferonγIL1β= interleukin 1betaIL33= interleukin 33IL37= interleukin 37LDL=lowdensity lipid (cholesterol)MCAS=mast cell activation syndromeMCP=monocyte chemoattractant proteinME/CFS=myalgic encephalomyelitis/chronic fatigue syndromeMetS=metabolic encephalomyelitisMI= myocardial infarctionMIF=macrophage inflammatory factorMiRNA= microRNAMIP=macrophage inflammatory protein mtDNA= mitochondrial DNA NGF=nerve growth factorNE=norepinephrinePTH= parathyroid hormonePDH=pyruvate dehydrogenasePDGF=plateletderived growth factorPPS/IC=Pelvic pain syndrome/Interstitial cystitisPoly (I:C)=polyinosinic:polycytidylic acidPOTS= Postural orthostatic tachycardia syndromePPAR=peroxisome proliferatoractivated receptorRANKL= Receptor activator of nuclear factor kappaΒ ligand ROS=reactive oxygen speciesSCF=stem cell factorSEID=systemic exertion intolerance diseaseSP= substance PTCA=tricarboxylic acidT2DM=Type 2 Diabetes MellitusTGFβ=transforming growth factor βTNF= tumor necrosis factorUCP2= uncoupling protein 2VEGF=vascular endothelial growth factor Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a complex disease characterized by debilitating fatigue, lasting for at least 6 months, with associated malaise, headaches, sleep disturbance and cognitive impairment, which severely impacts on quality of life. A significant percentage of ME/CFS patients remains undiagnosed, mainly due to the complexity of the disease and the lack of reliable objective biomarkers. ME/CFS patients display decreased metabolism and the severity of symptoms appears to be directly correlated to the degree of metabolic reduction that may be unique to each individual patient. However, the precise pathogenesis is still unknown preventing the development of effective treatments. The ME/CFS phenotype has been associated with abnormalities in energy metabolism, apparently due to mitochondrial dysfunction, in the absence of mitochondrial diseases, resulting in reduced oxidative metabolism, mitochondria may be further contributing to the ME/CSF symptomatology by extracellular secretion of mitochondrial DNA, which could act as an “innate” pathogen and create an autoinflammatory state in the hypothalamus. We propose that stimulation of hypothalamic mast cells by environmental neuroimmune pathogenic and stress triggers activates microglia leading to focal inflammation in the brain and disturbed homeostasis. This process could be targeted for the development of novel effective treatments.Myalgic Encephalomyelitits/Chronic Fatigue Syndrome (ME/CFS) is defined by the original diagnostic criteria (Fukuda, et al., 1994), and by the Canadian Consensus Criteria (Carruthers, et al., 2003), (Carruthers, 2007) followed by an international consensus (Carruthers, et al., 2011) and newer clinical diagnostic criteria developed by an NIH pathways to prevention workshop (Haney, et al., 2015) and the Institute of Medicine (Germain, et al., 2017). ME/CFS has also been known by other names (Unger, et al., 2016), most recently as Systemic Exertion Intolerance Disease (SEID),(Monro and Puri, 2018) ME/CFS is a complex disease that involves the muscular, nervous, hormonal and immune systems (Natelson, 2001),(Georgiades, et al., 2003), (Brurberg, et al., 2014), (Brigden, et al., 2017), (Scheibenbogen, et al., 2017). As the name implies, ME/CFS is characterized by debilitating fatigue lasting for at least 6 months, with severe impairment of daily functioning and associated symptoms, such as sleep disturbances, muscle aches, flulike malaise, gastrointestinal symptoms, orthostatic intolerance, chronic or intermittent pain, as well as cognitive impairment reflected as memory and concentration difficulties (Natelson, et al., 2007), {25039),(Yancey and Thomas, 2012), (Ganiats, 2015), (Komaroff, 2015), (Scheibenbogen, et al., 2017). The intensity of symptoms appears to be significantly affected by exertion (Rowe, et al., 2016). Anxiety and increased vulnerability to stress are also common in ME/CFS patients, including children affected by the disease (Smith, et al., 2003), (Crawley, et al., 2009). Abnormal hypothalamicpituitaryadrenal (HPA) axis activity has been observed in many patients (Cleare, et al., 2001), thus suggesting an association between ME/CFS and disturbed neuroendocrine mechanisms. Interestingly, ME/CFS patients are more likely to have migraine headaches than normal controls (Ravindran, et al., 2011). ME/CFS is often comorbid with disorders (Table 1) that are characterized by central nervous system (CNS) dysfunction, (MartinezMartinez, et al., 2014) and which are also negatively affected by stress (Theoharides and Cochrane, 2004), (Theoharides, 2013): Gulf War Illness (GWI) (Gwini, et al., 2016), Pelvic Pain Syndrome/Interstitial Cystitis (PPS/IC) (Whitmore and Theoharides, 2011), Fibromyalgia Syndrome (FMS) (Theoharides, et al., 2015c), and Mastocytosis (Theoharides, et al., 2015d) or Mast Cell activation syndrome (MCAS) (Petra, et al., 2015), (Akin, 2014). However, there are distinct differences between these other diseases such as between ME/CFS and FMS (Abbi and Natelson, 2013), (Pejovic, et al., 2015). ME/CFS is estimated to affect as many as 2.5 million people in the US, which corresponds to about 1% of the total US population. (Vincent, et al., 2012), (Komaroff, 2015), (Ganiats, 2015) Other studies (Jason, et al., 2009), including Minnesota (Vincent, et al., 2012), as well as from the UK (Nacul, et al., 2011), (Collin, et al., 2017), Norway (Bakken, et al., 2014) and Italy (Capelli, et al., 2015) report a lower incidence. Women are apparently more susceptible than men, with an estimated ratio of 4:1 (Germain, et al., 2017). The disease predominantly affects adults, even though symptoms may appear in childhood andadolescence (Crawley, 2014), (Nijhof, et al., 2011) ,(Jason, et al., 2006). Unfortunately, a significant number of suspected ME/CFS patients remain undiagnosed (Jason, et al., 2006) mainly due to the complexity of the disease and the lack of reliable diagnostic biomarkers (Klimas, et al., 2012). Multisystem diseases such as ME/CFS are often very timely and expensive to diagnose, and most patients go through years of searching and agony, as well as significant financial expenditures and impairment of their quality of life (Germain, et al., 2017). The economic health burden for ME/CFS in the USA was estimated to be $24 billion in 2018. (Jason, et al., 2008) . This makes imperative the need for the development of objective diagnostic biomarkers that will not only assist in the critical identification of patients with ME/CFS, but will also provide essential information on the pathophysiological mechanisms involved. A number of mechanisms and molecules have been implicated in the pathogenesis of ME/CFS(Gerwyn and Maes, 2017). Autoimmune (Sotzny, et al., 2018) and metabolic (Tomas and Newton,2018) pathways appear to play key roles in the pathophysiology of ME/CFS (Theoharides, et al.,2004b), (Maes, et al., 2011), (Booth, et al., 2012). Neuroimmune and neuroendocrine processes might also be involved, but are still largely unknown (Dietert and Dietert, 2008), (Bower, 2012). Clinical and subclinical viral infections have been suspected, but never confirmed, as a possible risk factor for the development of ME/CFS (Katz, et al., 2009), (Fremont, et al., 2009). The involvement of neuroinflammation of the brain has recently been suggested without any specific pathogenetic mechanism. (Glassford, 2017), (Tomas and Newton, 2018), (Morris, et al., 2018) Here we give an overview of the current understanding of the associations between ME/CFS and metabolic disease, and propose that focal inflammation in the hypothalamus due to local activation of mast cell and microglia, may alter homeostasis and provide a target for novel treatment approaches. Metabolic IrregularitiesME/CFS has been found to involve irregularities in the metabolism, energy, amino acid, nucleotide, nitrogen, hormone, and oxidative stress metabolism (Armstrong, et al., 2014), (Germain, et al., 2017). In particular, it has been proposed that the severe and prolonged fatigue experienced by ME/CFS patients may be a consequence of abnormalities in bioenergetic function (Tomas, et al., 2017). Much evidence suggests that the pathophysiology of ME/CFS is highly associated with alterations in normal energy metabolic processes (Fluge, et al., 2016) and abnormalities in cellular bioenergetics (Fluge, et al., 2016;Hornig, et al., 2015), (Fluge, et al., 2016), (Tomas, et al., 2017). There is also evidence to suggest that patients with ME/CFS might be at an increased risk for developing metabolic syndromeassociated diseases, such as diabetes, cardiovascular disease and thyroid disease (Maloney, et al., 2009). Apparently, systemic exertion intolerance in repeated cardiopulmonary exercise tests was demonstrated in ME/CFS patients present as compared to healthy controls suggesting insufficient metabolic adaptation to incremental exercise (Vermeulen and Vermeulen, I, 2014), (Keller, et al., 2014). It should be noted, that the Vermeulen and Vermeulen study including controls, which were not matched to ME/CFS in terms of fitness, while the Keller et al study had no controls. McCully et al published a number of papers showing that when matched for aerobic fitness, cardiorespiratory responses to exercise in patients with ME/CFS only and ME/CFS plus FM were not different from those in sedentary healthy controls (Cook, et al., 2006). Such intolerance, if real, may involve a switch to anaerobic glycolysis, i.e. a reduction in oxidative metabolism, and an increase in lactate production (Murrough, et al., 2010), (Shungu, et al., 2012b), which constitute the most common metabolic alterations observed in patients with ME/CFS. These characteristics have mainly been attributed to deconditioning, a state characterized by loss of muscle tone and power from prolonged lack of use (Bains, 2008). However, even though increased lactate production was originally noted, possibly related to the reduction of postexercise oxygen delivery (McCully, et al., 2004), the same effect could not be substantiated suggesting a possible decrease in oxygen delivery perhaps due to reduced blood flow (McCully and Natelson, 1999). In particular, there was elevated ventricular lactate, but no significant difference in high energy phosphatase metabolites in patients with ME/CFS as compared to patients with major depressive disorder or healthy volunteers (Shungu, et al., 2012a). In some cases, alterations in glucose utilization and lactate production were evident only after physical exercise of ME/CFS patients (Fluge, et al., 2016). ME/CFS plasma and serum metabolomics point in the direction of a hypometabolic state (Naviaux, et al., 2016), (Fluge, et al., 2016), (Germain, et al., 2017), (NagySzakal, et al., 2018). ME/CFS association with metabolic diseaseMetabolic syndrome (MetS) is a disorder characterized by an imbalance between energy expenditure and storage, and is diagnosed by the simultaneous presence of three of the following five conditions: (a) central type (or abdominal), (b) obesity, (c) increased blood pressure, elevated fasting glucose levels, (d) high levels of serum triglycerides, and (e) decreased highdensity lipid (HDL) cholesterol levels (Mottillo, et al., 2010), (Kaur, 2014). MetS is also linked to insulin resistance, a condition in which, despite normal insulin secretion by pancreatic βcells and hyperinsulinemia, can lead to hyperglycaemia and the development of Type II diabetes mellitus (T2DM) (Petersen and Shulman, 2006). In addition, high blood pressure and high cholesterol levels are closely linked to increased oxidative stress and endothelial dysfunction, thus enhancing the proinflammatory nature of microvascular atherosclerotic disease (Li, et al., 2007). In other words, subjects with MetS are at an increased risk of developing cardiovascular disease (CVD) and T2DM (Isomaa, et al., 2001), (Dekker, et al., 2005), (Petersen and Shulman, 2006). Approximately half of patients with ME/CFS also appear to have a previously undiagnosed medical condition, most often diabetes, CVD and thyroid diseases (Maloney, et al., 2009). Few studies have investigated the possible associations between MetS and ME/CFS (Maloney, et al., 2009), (Naviaux, et al., 2016), (Germain, et al., 2017), (Bozzini, et al., 2018). It was first suggested that patients with ME/CFS were twice as likely to have MetS, as compared to controls, after adjusting for bodymass index (BMI), waist circumference, triglycerides and glucose levels (Maloney, et al., 2009). MetS components in the ME/CFS group were significantly correlated with worse fatigue, but not with worse physical or mental functioning, contrary to previous observations (Tsai, et al., 2008), (Maloney, et al., 2009). A correlation of MetS with fatigue has also been observed in patients with FMS, a condition clinically similar to ME/CFS in which muscle pain and fatigue are the main symptoms; specifically, MetS components [lowdensity lipoprotein (LDL) cholesterol, as well as urinary norepinephrine (NE)/epinephrine and NE/cortisol rations], were significantly higher in women with FMS, as compared to healthy controls (Loevinger, et al., 2007). Some studies have reported abnormal findings concerning the cardiovascular system, but one study was in patients with small hearts (Miwa and Fujita, 2009;Azevedo, et al., 2007) and the other was in adolescents (Wyller, et al., 2008),and autonomic nervous system (ANS) dysfunction (Meeus, et al., 2013). Low blood pressure was noted in certain ambulatory cases of patients with ME/CFS (Newton, et al., 2009), (Wyller, et al., 2011), (Frith, et al., 2012). However, when patients with ME/CFS were matched to healthy controls by V02 max there were no differences in cardiovascular parameters (Cook, et al., 2006). Dysautonomia including Postural orthostatic tachycardia syndrome (POTS) may be present in many patients with ME/CFS (Hollingsworth, et al., 2010) and could also explain other ME/CFS symptoms, such as fatigue, vertigo, decreased concentration, tremors and nausea (Bozzini, et al., 2018). Interestingly, the low systolic blood pressure observed in ME/CFS patients is usually accompanied by exaggerated diurnal variation, which is inversely correlated with increasing fatigue (Davis, et al., 2000), (Newton, et al., 2009). Overall, it appears that metabolic disease components show significant correlations with the fatigue in ME/CFS patients and not with the disease itself. For example, blood pressure, as well as insulin resistance, are probably secondary to fatigue, and most probably reflect the lack of physical activity and prolonged lack of muscle use in ME/CFS patients. This makes sense if one considers that low blood pressure could give rise to fatigue through brain/or muscle hypoperfusion (Newton, et al., 2009), and that insulin sensitivity is highly dependent on the oxidative capacity of the muscle (Canto and Auwerx, 2009). Metabolomics, smallmolecule metabolite profiling (Daviss B., 2005), has provided relevant information that could distinguish ME/CFS patients (Naviaux, et al., 2016). Several studies have performed metabolite analysis of various biological fluids, [urine, blood, serum and cerebrospinal fluid (CSF)] from ME/CFS patients (Georgiades, et al., 2003), (Jones, et al., 2005), (Niblett, et al., 2007), (Suarez, et al., 2010), (Armstrong, et al., 2012), (Armstrong CW, et al., 2015), (Hornig, et al., 2016). However, despite confirming disturbances in energy, amino acid, nucleotide, nitrogen, hormone and oxidative stress metabolomics, they have not been able to determine a distinct, reproducible metabolic profile for ME/CFS (Germain, et al., 2017). Nevertheless, one study identified nine biochemical disturbances that were common to both male and female patients with ME/CFS, but not healthy controls (Naviaux, et al., 2016). Overall, there were marked decreases in sphingolipid, glycosphingolipid, phospholipid, purine, microbiome aromatic amino acid and branch chain amino acid metabolites, as well as in flavine adenine nucleotide (FAD) and lathosterol, which identified hypometabolic profile for ME/CFS. These changes correlated with disease severity and had an apparent diagnostic accuracy that exceeded 90% (Naviaux, et al., 2016). Interestingly, the metabolic abnormalities found in ME/CFS patients, were opposite (i.e. decreased instead of being increased), to those observed in MetS suggesting that ME/CFS patients could be more resistant to hypertension, dyslipidaemia, obesity and insulin resistance even though previous studies discussed above had reported an increased association between ME/CFS and metabolic syndrome.Another study that used targeted plasma metabolomics reported a similar trend of hypometabolic state in ME/CFS patients (Germain, et al., 2017). Even though the metabolite compounds were not all identical to the ones studied by Naviaux at al., both agreed on the presence of disturbances in lipid and fatty acid metabolism (Germain, et al., 2017). These findings are also in agreement with reported deficiencies in the urea and the TCA cycles, (ornithine/citrulline and pyruvate/isocitrate ratios), which ultimately result in reduced levels of ATP production in patients with ME/CFS (Yamano, et al., 2016). Other studies revealed that ME/CFS have reduced substrates that enter oxidation downstream of pyruvate dehydrogenase (PDH), such as glutamine, glutamate and phenylalanine, thus suggesting impaired pyruvate catabolism, which ultimately results in increased utilization of acetylCoAproducing amino acids as alternative substrates for fuelling aerobic metabolism via the TCA cycle (Armstrong, et al., 2012), (Armstrong CW, et al., 2015), (Fluge, et al., 2016). Reduced concentrations of amino acids that maintain TCA cycle capacity were detected in patients with ME/CFS (Fluge, et al., 2016), suggesting impaired fuelling of the TCA cycle by pyruvate. This finding is in line with the results of other studies where TCA cycle intermediates were also found to be reduced in both urine (Niblett, et al., 2007) and plasma (Yamano, et al., 2016) samples from ME/CFS patients. Mitochondrial dysfunctionOverall, the ME/CFS phenotype has been associated with mitochondrial dysfunction, 5' adenosine monophosphateactivated protein kinase (AMPK) impairment, oxidative stress and skeletal muscle cell acidosis (Myhill, et al., 2009), (Kennedy, et al., 2005), (Brown, et al., 2015), (Tomas, et al., 2017). The main ME/CFS symptoms, such as fatigue, exercise intolerance and myalgia, are also shared by patients diagnosed with primary mitochondrial disorders (Filler, et al., 2014), (Gorman, et al., 2015). However, unlike the mitochondrial dysfunction observed in mitochondrial disorders is known to be caused by mutations in either nuclear or mitochondrial DNA (mtDNA) (Tomas, et al., 2017), these mutations in patients with ME/CFS are extremely rare (BillingRoss, et al., 2016), (Schoeman, et al., 2017). In addition, certain mitochondrial enzymes have been found to discriminate between mitochondrial disorders and ME/CFS. Notably respiratory chain complex (RCC) I, III and IV activity (Smits, et al., 2011) appears to be significantly higher in ME/CFS patients. Instead, ATP production rate was found to be within the normal range in ME/CFS patients, but significantly decreased in approximately three quarters of the patients with mitochondrial disease, and was therefore regarded as the most reliable discrimination test (Smits, et al., 2011). Muscle biopsies from ME/CFS patients have shown mitochondrial degeneration, atrophy of type II fibers and fusion of mitochondrial cristae, decreased mitochondrial membrane permeability, severe deletions in mtDNA genes that are involved in cellular energy processes, as well as oxidative damage from increased production of free radicals (Myhill, et al., 2009), (Morris and Maes, 2013). Mitochondrial dysfunction has also been observed in peripheral mononuclear blood cells (PMBC) of ME/CFS patients, even though it has not yet been elucidated if they constitute the cause of the disease (Myhill, et al., 2009), (Myhill, et al., 2013), (Tomas, et al., 2017). Notably, a significant correlation has been observed between the extent of mitochondrial dysfunction and the degree of ME/CFS severity, thus suggesting that mitochondrial dysfunction might be a contributing factor in ME/CFS pathology, at least in a subset of patients (Myhill, et al., 2009), (Booth, et al., 2012). However, it is difficult to assess mitochondrial dysfunction that is usually done by measuring the levels of lactate and pyruvate in the serum, best done by serial serum sampling from an arm after a brief period of exercise. When limited amounts of oxygen are available, as is usually the case with intense exercise, anaerobic glycolysis, or otherwise called the lactic acid system, provides an effective means of energy production. During this process, glucose is catabolized via the glycolytic pathway, resulting in pyruvate being converted to lactate by lactate dehydrogenase. This process lasts 1030 seconds during maximal effort and produces about 5% of the glucose energy potential in the form of adenosine5´triphosphate (ATP) molecules (2 molecules of ATP for every molecule of glucose). ATP synthesis can be estimated by measuring the anaerobic threshold (AT), i.e. the rate of oxygen consumption at work rate when blood lactic acid begins to accumulate, and the maximal work rate (Morris and Maes, 2014). The AT indicates a switch during which ATP synthesis stops being produced by mitochondria and occurs via the anaerobic route (Morris and Maes, 2012), whereas anaerobic threshold and recovery time following exercise depends on lactate production and clearance rates (Fluge, et al., 2016). When aerobic conditions are normal, pyruvate is transported into mitochondria and converted to acetylCoA by either PDH or via degradation of fatty acids and ketogenic amino acids. In either case, acetylCoA is further oxidized in the tricarboxylic acid (TCA) cycle, producing some ATP, and the electron transport chain (respiratory chain), which generates ATP from ADP by oxidative phosphorylation (oxphos). AcetylCoA thereby serves to fuel mitochondrial respiration and ATP production by oxidative phosphorylation (Fluge, et al., 2016) for essential tissue functions (Myhill, et al., 2009). Reduced ATP production is associated with increased levels of reactive oxygen species (ROS), which may ultimately lead to mitochondrial damage and the hypometabolic profile of ME/CFS (Naviaux, et al., 2016), (Armstrong CW, et al., 2015). Severely reduced or impaired mitochondrial oxidative phosphorylation in ME/CFS patients is highly correlated with significantly increased intracellular lactate levels, even in the recovery phase of a mild exercise where ATP synthesis is extremely low (Vermeulen, et al., 2010), (Morris and Maes, 2014). Among the factors that may contribute to mitochondrial dysfunction, the most prominent ones appear to be increased levels of proinflammatory cytokines, such as interleukin1beta (IL1β) and tumor necrosis factor (TNF), which directly inhibit mitochondrial respiration by increasing mitochondrial membrane permeability, which ultimately leads to membrane depolarization and an increased production of ROS (Morris and Maes, 2013). However, even though TNF is elevated in the serum of patients with FMS, (Theoharides, et al., 2010c) it was not consistently elevated in ME/CFS (Brenu, et al., 2011), but was apparently associated only with increased IL4 (Hanson, et al., 2001).There was also no significant difference in serum cytokine levels across the night (Nakamura, et al., 2010) or post exercise (Nakamura, et al., 2013). There is some evidence of stronger correlation of cytokines alterations early in the course of illness rather than severity (Hornig, et al., 2015). It has been proposed that “cytokine coexpression networks” may be more predictive of ME/CFS phenotype (Klimas, et al., 2012), (Hornig, et al., 2016), but looking for such biomarkers in the periphery would not reflect inflammation in the brain. One study reported that of 27 cytokines studied in CSF from ME/CFS patients, only IL10 was significantly reduced {26107}. Another paper using network analysis of CSF cytokine levels reported an inverse relationship with interleukin 1 receptor antagonist only in classical, but not in atypical ME/CFS (Hornig, et al., 2017). Certain microRNAs (miRNAs) may turn out to be distinct or differentially expressed in ME/CFS. Recently, miRNAs have been implicated in the hypothalamic control of energy homeostasis (Najam, et al., 2018). However, the available studies in patients with ME/CFS did not report any consistent pattern whether pre\ or postexercise, plasma,(Brenu, et al., 2014) NK cells (Petty, et al., 2016) or CD8+ cells (Brenu, et al., 2012). One recent important study showed exercise induced changes in CSF fluid from patients with ME/CFS, Gulf War Illness and sedentary controls found twelve diminished miRNAs after exercise (Baraniuk and Shivapurkar, 2017), (Baraniuk and Shivapurkar, 2018). Focal Inflammation in the Diencephalon and Dysfunctional HPA axisNeuroinflammation (Nakatomi, et al., 2014), (Glassford, 2017), (Tomas and Newton, 2018), (Morris, et al., 2018) and immune dysfunction (Morris, et al., 2014), (Nijs, et al., 2014), (Trivedi, et al., 2018) have been suggested as being involved in the pathogenesis of ME/CFS, but serum levels of proinflammatory cytokines have not been confirmed as discussed later. Considerable evidence indicates that ME/CFS is characterized by dysfunction of the HPA axis, (Theoharides, et al., 2010b), (Morris, et al., 2016) and symptoms are known to worsen by stress (Smith, et al., 2003)), (Theoharides and Cochrane, 2004), ((Crawley, et al., 2009;Theoharides and Cochrane, 2004;Theoharides, 2013). Stress can also worsen or precipitate obesity and cardiovascular events (Theoharides, et al., 2008), (Theoharides, et al., 2011), (Alevizos, et al., 2013), (Sismanopoulos, et al., 2013), through local inflammation (Matusik, et al., 2012;Libby, et al., 2002). Corticotropinreleasing hormone (CRH) is secreted from the hypothalamus under stress and stimulates the HPA axis via activation of two main types of G proteincoupled receptors, CRHR1 and CRHR2 (Chrousos, 1995). CRH secreted under acute stress, has been implicated in the pathophysiology of neuroinflammatory disorders and myocardial infarction (MI) (Jiang, et al., 1996;Krantz, et al., 2000;O'Kane, et al., 2006;Slominski, 2009). We propose that stimulation of hypothalamic mast cells by environment, neural, immune pathogenic (Lyme, mycotoxins) or stress triggers (CRH, somatostatin) activates microglia leading to focal inflammation and disturbed homeostasis (Figure 1). Mast cell and/or microglia triggers may derive from the nasal cavity, or may reach the brain area through a disrupted BBB or through the lymphatics. Stimulated mast cells could secrete molecules that can alter homeostasis directly (via secretion of CRH, urocortin) or activate microglia (via secretion of histamine, tryptase and mtDNA). Microglia then release more inflammatory molecules (IL1β, IL6, and CCL2) that further disrupt homeostasis, causes mitochondrial dysfunction and contribute to fatigue both centrally and peripherally. In fact, activated microglia have been reported to contribute to the pathophysiology of sleep disorders (Nadjar, et al., 2017). The involvement of more than one trigger can lead to a significantly heightened response and lower the triggering threshold of both mast cells and microglia leading to chronic symptoms. Mast cells are unique tissue immune cells involved in allergic reactions (Theoharides, et al., 2015d), but also act as sensors of environmental and psychological stress (Theoharides, 2017). Even though we invoke stimulation of mast cells in the hypothalamus, it does not necessarily mean that mast cells should necessarily be stimulated outside the CNS. Nevertheless, there have been reports of an association between ME/CFS and acute rhinitis including significantly higher TNF and CXCL8 levels in nasal lavage fluid (RepkaRamirez, et al., 2002). In addition, chronic rhinosinusitis symptoms were significantly higher in patients with ME/CFS (Chester, 2003), apparently due to nonallergic rhinitis (Baraniuk and Ho, 2007). It is well known that both allergic and perennial rhinitis involve activation of mast cells (Bachert, et al., 2018). More recently, it was reported that the incidence of ME/CFS was higher in patients with a history of atopy (Yang, et al., 2015). Moreover, circulating blood mast cell precursors were found to be higher in ME/CFS patients (Nguyen, et al., 2017).Mast cells are located perivascularly in the hypothalamus, thalamus and third ventricle of the diencephalon (Edvinsson, et al., 1977), (Pang, et al., 1996). CRH could stimulate MC in the hypothalamus since CRHR1 gene is expressed on human cultured mast cells, activation of which induces production of vascular endothelial growth factor (VEGF), (Cao, et al., 2005) which could increase permeability of the bloodbrain barrier (BBB) (Theoharides and Konstantinidou, 2007), (Theoharides, 1990), (Esposito, et al., 2002) leading to inflammation of the brain (Theoharides, et al., 2004a). Moreover, CRH is synthesized by mast cells (Kempuraj, et al., 2004) implying it could have autocrine effects. Interestingly, even somatostatin stimulates mast cells (Theoharides, et al., 1990). Mast cells are also found in the pineal, the pituitary and the thyroid glands (Theoharides, 2017) further extending their contribution to the symptoms of ME/CFS such as sleep disturbances dysfunctional HPA axis and fatigue due to thyroid dysfunction. Mast cells are wellknown for their role in allergic reactions, (Beaven, 2009) but mast cells are now considered important in innate and acquired immunity, (Galli, et al., 2008) antigen presentation, (Gong, et al., 2010) and inflammation (Theoharides, et al., 2010a). Mast cells can be stimulated by neurons, hormones, environmental, neuroimmune, pathogenic and stress triggers. (Table 3), (Theoharides, et al., 2015d), (Theoharides, 2017). Reactive oxygen species (ROS) can also stimulate mast cells (Swindle and Metcalfe, 2007). (Robuffo, et al., 2017), (Toniato, et al., 2017) Mast cells also secrete leptin that could contribute to cachexia and fatigue (Taildeman, et al., 2009). Mast cells secrete as many as 100 different mediators (Table 4) (Mukai, et al., 2018), (Theoharides and Kalogeromitros, 2006) (Wernersson and Pejler, 2014) often selectively without degranulation (Theoharides, et al., 2007), utilizing different secretory pathways (Xu, et al., 2018). Mast cells can also secrete danger signals, (Theoharides, 2016), including many chemokines and cytokines (Conti, et al., 2017),(Mukai, et al., 2018) especially mitochondrial DNA (mtDNA), (Zhang, et al., 2012) which could act as an “innate pathogen” (Zhang, et al., 2011) leading to a localized brain autoinflammatory response (Collins, et al., 2004;Marques, et al., 2012;Sun, et al., 2013;Theoharides, et al., 2013). Extracellular mtDNA could either be secreted directly in the diencephalon or could reach the brain through lymphatics (Louveau, et al., 2015). We had reported that mtDNA is increased in the serum of children with autism spectrum disorder (ASD) (Zhang B, et al., 2010). Mast cellderived mediators can then stimulate microglia (Zhang, et al., 2016), (Patel, et al., 2016) to secrete additional proinflammatory and homeostasisdisrupting molecules (Table 5) contributing to fatigue and neuropsychiatric symptoms (Theoharides TC., et al., 2016). It is interesting that peptide Y was found to be elevated in plasma of patients with ME/CFS and correlated significantly with stress (Fletcher, et al., 2010), as this peptide is known to stimulate mast cells (Mousli and Landry, 1994). An important part is that combination of triggers is likely to play a more important pathogenetic role than individual ones. For instance, we reported that combination of CRH and NT have synergistic action in stimulating VEGF secretion without tryptase from human mast cells (Donelan, et al., 2006), as well as induce the expression of each other’s receptors on human mast cells (Alysandratos, et al., 2012). More recently, we showed that the combination of SP and IL33 has synergistic action in stimulating TNF secretion without tryptase from human cultured mast cells (Taracanova, et al., 2017c). CRH is often released together with another peptide, neurotensin (NT), which is vasoactive (Leeman and Carraway, 1982) and has also been implicated in inflammation (Mustain, et al., 2011) and neurological diseases (Caceda, et al., 2006). NT is increased in the skin following acute stress (Theoharides, et al., 1998) and increases vascular permeability, an effect synergistic with CRH (Crompton, et al., 2003), (Donelan, et al., 2006). Mast cells are also stimulated by the peptide Substance P (SP), (Church, et al., 1991;Theoharides, et al., 2010d;Taracanova, et al., 2017a) initially characterized by Leeman and colleagues, (Chang and Leeman, 1970;Carraway and Leeman, 1973) and shown to participate in inflammatory processes (Mashaghi, et al., 2016;O'Connor, et al., 2004;Hokfelt, et al., 2001;Douglas and Leeman, 2011). IL33 is a member of the IL1 family of cytokines and has emerged as an early warning sign (dubbed “alarmin”) (Moulin, et al., 2007) in autoimmune or inflammatory process (Saluja, et al., 2015;Theoharides, et al., 2015a;Theoharides, 2016). IL33 is secreted by fibroblasts and endothelial cells, (Liew, et al., 2010) but also from mast cells. (Tung, et al., 2014) IL33 augments the effect of IgE on secretion of histamine from mast cells and basophils (Moulin, et al., 2007), (Silver, et al., 2010), but the effect of IL33 when used by itself or in combination with SP on secretion of IL1β from human mast cells has not been reported. Substance P stimulated secretion of VEGF, an action augmented by IL33 (Theoharides, et al., 2010e). We recently showed that stimulation of human mast cells by SP given together with IL33 markedly increases secretion and gene expression of the proinflammatory cytokine, TNF (Taracanova, et al., 2017b). Interestingly, chronic rhinosinusitis, which is quite common in patients with ME/CFS as discussed earlier, has been associated with high levels of nasal IL33 (Ozturan, et al., 2017), which could reach the hypothalamus through the cribriform plexus. Does any treatment modality work?There are currently no FDA approved drugs for the treatment of ME/CFS and the available psychological, physical and pharmacological interventions do not appear to be effective (Bains, 2008;Pae, et al., 2009;Morris and Maes, 2014;Loades, et al., 2016;Collatz, et al., 2016;CastroMarrero, et al., 2017;Brigden, et al., 2017). Mitochondria appear as one appealing drug target for the treatment of ME/CFS, but other papers reported no apparent alteration in ATP production (Shungu, et al., 2012b). Chemokines and cytokines have been proposed as targets for neuroinflammatory disorders (Pranzatelli, 2018), but such have not been tried in ME/CFS.The peroxisome proliferatoractivated receptor (PPAR) agonist bezafibrate improves mitochondrial function by stimulating mitochondrial biogenesis and increasing the oxidative phosphorylation efficiency in a number of studies (Valero, 2014;Wang, et al., 2010;Johri, et al., 2012). It has also been suggested that, since fatigue is associated with hypotension in ME/CFS patients, increasing blood pressure might present an effective therapeutic approach to this symptom. Even though previous studies using the mineralcorticoid fludrocortisone failed to show any improvement (Peterson, et al., 1998), (Rowe, et al., 2016), use of the agonist midodrine to increase blood pressure has produced some improvement of the fatigue (Naschitz, et al., 2004). Interestingly, angiotensin II inhibitors have been shown to increase mitochondrial membrane potential, to improve mitochondrial function and to stimulate mitochondrial biogenesis (Morris and Maes, 2014), (de Cavanagh, et al., 2011). Indeed, blockade of angiotensin II has been shown to prevent the onset of T2DM in mice by increasing fat oxidation, decreasing muscle triglycerides and improving glucose tolerance (Mitsuishi, et al., 2009). The angiotensin receptor blocker telmisartan improves mitochondrial dysfunction by enhancing mitochondrial biogenesis and protecting vascular and endothelial cell damage (Takeuchi, et al., 2013), (Kurokawa, et al., 2015). Similarly, the angiotensin receptor blocker losartan has been shown to improve mitochondrial respiratory chain function and coenzyme Q10 (CoQ10) content in hypertensive animals (Sumbalova, et al., 2010). However, given the blood pressure lowering effects of these agents it is unlikely they will be useful in ME/CFS, except maybe in select patients. Several natural compounds may have a beneficial effect on mitochondrial function. Magnesium ions play critical roles in energy metabolism and in maintaining normal muscle function, by being positively active regulator of glycolysis and of all enzymatic reactions involving phosphate group transfer from ATP (Dominguez, et al., 2006), (Morris and Maes, 2014). Several studies have demonstrated that magnesium ion supplements significantly increase muscle strength and maintain optimal physical activity performance in humans (Brilla and Haley, 1992), (Newhouse and Finstad, 2000), (Kass and Poeira, 2015), (Zhang, et al., 2017). In experimental animals, this improvement in exercise performance seems to occur via enhancing glucose availability in the brain and muscle, and via reducing/delaying lactate accumulation (Zhang, et al., 2017). Magnesium sulphate may also improve mitochondrial respiratory function and prevent nitrous oxide production in the brain (Xu, et al., 2002), (Yang X, et al., 2007). Coenzyme Q10 deficiency has been reported in patients with ME/CFS (Maes, et al., 2009), (Maes, et al., 2012), (Filler, et al., 2014). However, administration of CoQ10 to patients with ME/CFS have failed to show any benefit (Campagnolo, et al., 2017). Naturally occurring flavonoids have potent antioxidant, antiinflammatory and neuroprotective actions (Guo, et al., 2009;Middleton, et al., 2000;Xiao, et al., 2011) and are generally considered safe (Harwood, et al., 2007;Kawanishi, et al., 2005;Theoharides, et al., 2014;Theoharides, et al., 2014). The flavonoid genistein, attenuates muscle fatigue in humans by downregulating oxidative stress and enhancing antioxidant enzyme activity (Ding and Liu, 2011). The flavonoids epigallocatechin, naringin and curcumin can ameliorate ME/CFS symptoms in experimental models (Sachdeva, et al., 2009), (Vij, et al., 2009), (Gupta, et al., 2009), (Sachdeva, et al., 2011). Other reports have documented similar chronic fatigue attenuating effects for the Astragalus flavonoids (Kuo, et al., 2009) and of olive extract (Gupta, et al., 2010). The isoflavones genistein and daidzein, have been shown to reverse the effects of polyinosinic:polycytidylic acid (poly(I:C) on mouse locomotor activity and brain inflammatory mediator expression in a mouse model of fatigue (Vasiadi, et al., 2014). Quercetin appears to increase exercise tolerance by attenuating oxidative stress in mouse brain, while at the same time conferring antioxidant and antiinflammatory action (Kempuraj, et al., 2005), (Davis, et al., 2009), (Ishisaka, et al., 2011). Luteolin suppresses adipocyte activation of macrophages and inflammation (Deqiu, et al., 2011;Ando, et al., 2009), while it increases insulin sensitivity of the endothelium (Deqiu, et al., 2011). Luteolin also inhibits mast cells (Asadi, et al., 2010;Weng, et al., 2015;Patel and Theoharides, 2017) and microglia (Jang, et al., 2008),(Patel, et al., 2016). In this context, it is interesting that luteolin improved symptoms of both ASD (Taliou, et al., 2013), (Tsilioni, et al., 2015), postLyme syndrome (Theoharides and Stewart, 2016) and brain fog (Theoharides, et al., 2015b) in openlabel trials. We recently showed that tetramethoxyluteolin is more potent than luteolin in its ability to inhibit human cultured microglia (Patel, et al., 2016) and mast cells (Patel and Theoharides, 2017). Intranasal administration of select flavonoids may reduce inflammation in the hypothalamus and correct the central pathogenesis of ME/CFS. Novel treatment approaches are required to address the central pathogenic processes. For instance, intranasal administration of microvesicleentrapped curcumin was shown to inhibit inflammation of the brain in a mouse model (Sun, et al., 2010). ConclusionsOverall, the ME/CFS phenotype has been associated with apparent abnormalities in the metabolic profile, possibly due to local inflammation in the hypothalamus. Compounds that could inhibit inflammation in the brain, such as tetramethoxyluteolin or the antiinflammatory cytokine IL37 (Dinarello, et al., 2016), (Mastrangelo, et al., 2018), may be potential treatment options. DISCLOSURESTCT is the inventor of US patents No. 7,906,153; No. 8,268,365 and PCT application No. 13/722, 397 for the treatment of neuroinflammatory conditions. Diagrammatic representation of the proposed mast cellmicroglia interactions in the hypothalamus, contribute to the pathogenesis of ME/CFS, and could serve as targets for treatment.Hypothalamic mast cells are stimulated by stressassociated triggers such as CRH, HK1 and SP along with mtDNA and IL33, some derived from nasal cavity, while others may reach the area through a disrupted bloodbrain barrier or through lymphatics. Stimulated mast cells then secrete molecules such as CXCL8, NT, TNF, tryptase and mtDNA, (CXCL) which activate microglia to secrete more inflammatory molecules especially, IL1β, IL6, and CXCL8 that further disrupt homeostasis, causes mitochondrial dysfunction and contribute to symptoms of ME/CFS. Luteolin could inhibit these processes at different steps as shown. Table 1. Conditions Often Comorbid with ME/CFS\_______________________________________________________________________\_• Chronic inflammatory response syndrome (CIRS)• Fibromyalgia syndrome (FMS)• EhlersDanlos Syndrome (EDS)• Gulf War Illness (GWI)• Interstitial cystitis/bladder pain syndrome (IC/BPS)• Irritable bowel syndrome (IBS)• Mast cell activation syndrome (MCAS)• Multiple chemical sensitivity syndrome (MCSS)• PostLyme syndrome• Postural orthostatic tachycardia syndrome (POTS)• Posttraumatic stress disorder (PTSD)• Restless leg syndrome\______________________________________________________________________\_ Table 2. Dysregulated Molecules that May Contribute to the Pathogenesis of ME/CFS \______________________________________________________________________\_ • Cachexins• Calcineurin• Heavy metals• Herbicides• Inflammatory cytokines• Leptin• Melatonin• miRNAs• Mitochondrial enzymes• Neuroendocrine disruptors• Neuropeptides• Neurotransmitters• Reactive Oxygen Species• Toxins (mycotoxins, Borrelia toxins)• Uncoupling protein 2• Xenobiotics\______________________________________________________________________\_ Table 3. Mast Cell Triggers Stimulating degranulation AcetylcholineAdenosineComplement fragments* C3α, C4α, C5αDrugs* Local anesthetics, lactam antibiotics, neuromuscular junction blockers, vancomycinEosinophil granule proteinsIgEIgG1IgG4LysophosphatidylserineHistamineSerotoninLysophosphatidic acidPeptides* Adrenomedullin, CGRP, Endorphin, Endothelin, Hemokinin1, Leptin, Mastoparan,Neurotensin, NGF, PTH, Somatostatin, SP, Thrombin, VIPTryptase Stimulating selective release of mediators without degranulation ATPBorrelia burgdorferi (Lyme Disease)CRHHeavy metals* Aluminum, cadmium, mercuryHerbicides* Atrazine, glyphosateIL33MycotoxinsLPSSCFViruses _\__\__________________________________________________________________\_ Table 4. Mast Cell MediatorsMediators Pathophysiologic effect PrestoredBiogenic AminesDopamine NeurotransmissionHistamine Vasodilation, angiogenesis, mitogenesis, pain5Hydroxytryptamine (5HT, serotonin) Polyamines Vasoconstriction, painSpermidine, spermine Secretory granule stability, inhibition of secretionChemokinesIL8 (CXCL8), MCP1 (CCL2), MCP3 (CCL7), Chemoattraction and tissue infiltration of leukocytes MCP4, RANTES (CCL5), Eotaxin (CCL11) CytokinesIL4, IL5, IL6, IL15, IL17, IL31, IL33, TNFEnzymes Immune cell maturation, inflammationArylsulfatases A Lipid/proteoglycan hydrolysisBetahexosaminidase Degradation processesBetaglucuronidase Degradation processesBetaglucosaminidase Degradation processesBetaDgalactosidase Degradation processesCarboxypeptidase A Peptide processingCathepsins B,C, D, E, L Degradation processesChymase Tissue damage, pain, angiotensin II synthesisGrnzyme B Inflammation and preapoptotic effectsKinogenases Synthesis of vasodilatory kinins, painPhospholipases Arachidonic acid generationRenin Angiotensin II generationTryptase Tissue damage, activation of PAR, inflammation, painMetalloproteinases(CPA3, MMP9, ADAMTSS)Growth factors Tissue damage, modification of cytokines/chemokinesbFGF NeovascularizationNGF Nerve growth, mast cell activationSCF Mast cell growth and activationTGFβ Antiinflammatory, profibroticVEGF PeptidesACTH Neovascularization, vasodilationAngiogenin NeovascularizationAngiopoietin NeovascularizationCorticotropinreleasing hormone Inflammation, mast cell stimulus, vasodilationEndorphins AnalgesiaEndothelin SepsisHemokinin1 Inflammation, mast cell stimulus, pain, vasodilationKinins (bradykinin) Inflammation, mast cell stimulus, pain, vasodilationLeptin Food intake regulatorMeltonin Biologic clock regulatorNeurotensin Inflammation, mast cell stimulus, vasodilationRANKL Osteoclast differentiation and activationSomatostatin Mast cell stimulant, antisecretorySubstance P Inflammation, mast cell stimulus, painUrocortin Inflammation, vasodilationVasoactive intestinal peptide Proteoglycans Vasodilation, mast cell activationChondroitin sulfate Cartilage synthesis, antiinflammatoryHeparin Angiogenesis, nerve growth factor stabilizationHyaluronic acid Connective tissue, nerve growth factor stabilizationSerglycin De novo synthesizedChemokinesCCL2, CXCL8, MIP1α, MCP1Cytokines Storage of granule proteasesInterleukins (IL)\1,2,3,4,5,6,8,9,10,13,16,18 Inflammation, leukocyte migration, painIFNα, IFN\ β, IFNγ; MIF; TGFβ; TNF, Growth Factors Inflammation, leukocyte proliferation/activationSCF, βFGF, neurotrophin 3, NGF, Growth of a variety of cellsPDGF, TGFβ, VEGFNitric oxidePhospholipid metabolites VasodilationLeukotriene B4 Leukocyte chemotaxisLeukotriene C4 Vasoconstriction, painPlatelet activating factor Platelet activation, vasodilationProstaglandin D2 Bronchonstriction, pain \______________________________________________________________________\_Table 5. Microglia Mediators\____________________________________________________________________\_ Cytokines• IL1β• IL6• TNF Chemokines• CCL2• CXCL8 (IL8)• CCL5 (MCP1)\____________________________________________________________________\_ Figure 1. Much evidence suggests that the pathophysiology of ME/CFS is highly associated with alterations in normal energy metabolic processes (Fluge, et al., 2016) and abnormalities in cellular bioenergetics (Fluge, et al., 2016;Hornig, et al., 2015), (Fluge, et al., 2016), (Tomas, et al., 2017) REFERENCES Abbi B and Natelson BH (2013) Is chronic fatigue syndrome the same illness as fibromyalgia: evaluating the 'single syndrome' hypothesis. QJM 106:39.Akin C (2014) Mast Cell Activation Disorders. J Allergy Clin Immunol Pract 2:252257.Alevizos M, Karagkouni A, Panagiotidou S, Vasiadi M and Theoharides TC (2013) Stress triggers coronary mast cells leading to cardiac events. Ann Allergy Asthma Immunol 112:309316.Alysandratos KD, Asadi S, Angelidou A, Zhang B, Sismanopoulos N, Yang H, Critchfield A and Theoharides TC (2012) Neurotensin and CRH interactions augment human mast cell activation. PloS One 7:e48934.Ando C, Takahashi N, Hirai S, Nishimura K, Lin S, Uemura T, Goto T, Yu R, Nakagami J, Murakami S and Kawada T (2009) Luteolin, a foodderived flavonoid, suppresses adipocytedependent activation of macrophages by inhibiting JNK activation. FEBS Lett 583:36493654.Armstrong CW, McGregor NR, Lewis DP, Butt HL and Gooley PR. (2015) Metabolic profiling reveals anomalous energy metabolism and oxidative stress pathways in chronic fatigue syndrome patients. Metabolomics 11: 16261639.Armstrong CW, McGregor NR, Butt HL and Gooley PR (2014) Metabolism in chronic fatigue syndrome. Adv Clin Chem 66:121172.Armstrong CW, McGregor NR, Sheedy JR, Buttfield I, Butt HL and Gooley PR (2012) NMR metabolic profiling of serum identifies amino acid disturbances in chronic fatigue syndrome. Clin Chim Acta 413:15251531.Asadi S, Zhang B, Weng Z, Angelidou A, Kempuraj D, Alysandratos KD and Theoharides TC (2010) Luteolin and thiosalicylate inhibit HgCl(2) and thimerosalinduced VEGF release from human mast cells. Int J Immunopathol Pharmacol 23:10151020.Azevedo A, Bettencourt P, Pimenta J, Frioes F, breuLima C, Hense HW and Barros H (2007) Clinical syndrome suggestive of heart failure is frequently attributable to noncardiac disorderspopulationbased study. Eur J Heart Fail 9:391396.Bachert C, Zhang N, Hellings PW and Bousquet J (2018) Endotypedriven care pathways in patients with chronic rhinosinusitis. J Allergy Clin Immunol 141:15431551.Bains W (2008) Treating Chronic Fatigue states as a disease of the regulation of energy metabolism. Med Hypotheses 71:481488.Bakken IJ, Tveito K, Gunnes N, Ghaderi S, Stoltenberg C, Trogstad L, Haberg SE and Magnus P (2014) Two age peaks in the incidence of chronic fatigue syndrome/myalgic encephalomyelitis: a populationbased registry study from Norway 20082012. BMC Med 12:167.Baraniuk JN and Ho LU (2007) The nonallergic rhinitis of chronic fatigue syndrome. Clin Allergy Immunol 19:427447.Baraniuk JN and Shivapurkar N (2017) Exercise induced changes in cerebrospinal fluid miRNAs in Gulf War Illness, Chronic Fatigue Syndrome and sedentary control subjects. Sci Rep 7:15338.Baraniuk JN and Shivapurkar N (2018) Author Correction: Exercise induced changes in cerebrospinal fluid miRNAs in Gulf War Illness, Chronic Fatigue Syndrome and sedentary control subjects. Sci Rep 8:6455.Beaven MA (2009) Our perception of the mast cell from Paul Ehrlich to now. Eur J Immunol 39:1125.BillingRoss P, Germain A, Ye K, Keinan A, Gu Z and Hanson MR (2016) Mitochondrial DNA variants correlate with symptoms in myalgic encephalomyelitis/chronic fatigue syndrome. J Transl Med 14:19.Booth NE, Myhill S and LarenHoward J (2012) Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int J Clin Exp Med 5:208220.Bower JE (2012) Fatigue, brain, behavior, and immunity: summary of the 2012 Named Series on fatigue. Brain Behav Immun 26:12201223.Bozzini S, Albergati A, Capelli E, Lorusso L, Gazzaruso C, Pelissero G and Falcone C (2018) Cardiovascular characteristics of chronic fatigue syndrome. Biomed Rep 8:2630.Brenu EW, Ashton KJ, Batovska J, Staines DR and MarshallGradisnik SM (2014) Highthroughput sequencing of plasma microRNA in chronic fatigue syndrome/myalgic encephalomyelitis. PLoS ONE 9:e102783.Brenu EW, Ashton KJ, van DM, Staines DR, Peterson D, Atkinson GM and MarshallGradisnik SM (2012) Cytotoxic lymphocyte microRNAs as prospective biomarkers for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J Affect Disord 141:261269.Brenu EW, van Driel ML, Staines DR, Ashton KJ, Ramos SB, Keane J, Klimas NG and MarshallGradisnik SM (2011) Immunological abnormalities as potential biomarkers in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J Transl Med 9:81.Brigden A, Loades M, Abbott A, BondKendall J and Crawley E (2017) Practical management of chronic fatigue syndrome or myalgic encephalomyelitis in childhood. Arch Dis Child 102:981986.Brilla LR and Haley TF (1992) Effect of magnesium supplementation on strength training in humans. J Am Coll Nutr 11:326329.Brown AE, Jones DE, Walker M and Newton JL (2015) Abnormalities of AMPK activation and glucose uptake in cultured skeletal muscle cells from individuals with chronic fatigue syndrome. PLoS ONE 10:e0122982.Brurberg KG, Fonhus MS, Larun L, Flottorp S and Malterud K (2014) Case definitions for chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME): a systematic review. BMJ Open 4:e003973.Caceda R, Kinkead B and Nemeroff CB (2006) Neurotensin: role in psychiatric and neurological diseases. Peptides 27:23852404.Campagnolo N, Johnston S, Collatz A, Staines D and MarshallGradisnik S (2017) Dietary and nutrition interventions for the therapeutic treatment of chronic fatigue syndrome/myalgic encephalomyelitis: a systematic review. J Hum Nutr Diet 30:247259.Canto C and Auwerx J (2009) PGC1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20:98105.Cao J, Papadopoulou N, Kempuraj D, Boucher WS, Sugimoto K, Cetrulo CL and Theoharides TC (2005) Human mast cells express corticotropinreleasing hormone (CRH) receptors and CRH leads to selective secretion of vascular endothelial growth factor. J Immunol 174:76657675.Capelli E, Lorusso L, Ghitti M, Venturini L, Cusa C and Ricevuti G (2015) Chronic fatigue syndrome: Features of a population of patients from northern Italy. Int J Immunopathol Pharmacol 28:5359.Carraway R and Leeman SE (1973) The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J Biol Chem 248:68546861.Carruthers BM (2007) Definitions and aetiology of myalgic encephalomyelitis: how the Canadian consensus clinical definition of myalgic encephalomyelitis works. J Clin Pathol 60:117119.Carruthers BM, Jain AK, DE Meirleir KL, Peterson DL, Klimas NG, Lerner AM, Bested AC, FlorHenry P, Joshi P, Powles ACP, Sherkey JA and van de Sande MI (2003) Myalgic encephalomyelitis/chronic fatigue syndrome: clinical working case definition, diagnostic and treatment protocols. Journal of Chronic Fatigue Syndrome 11(1).Carruthers BM, van de Sande MI, DE Meirleir KL, Klimas NG, Broderick G, Mitchell T, StainesD, Powles AC, Speight N, Vallings R, Bateman L, BaumgartenAustrheim B, Bell DS, CarloStella N, Chia J, Darragh A, Jo D, Lewis D, Light AR, MarshallGradisbik S, Mena I, Mikovits JA, Miwa K, Murovska M, Pall ML and Stevens S (2011) Myalgic encephalomyelitis: International Consensus Criteria. J Intern Med 270:327338.CastroMarrero J, SaezFrancas N, Santillo D and Alegre J (2017) Treatment and management of chronic fatigue syndrome/myalgic encephalomyelitis: all roads lead to Rome. Br J Pharmacol 174:345369.Chang MM and Leeman SE (1970) Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. J Biol Chem 245:47844790.Chester AC (2003) Symptoms of rhinosinusitis in patients with unexplained chronic fatigue or bodily pain: a pilot study. Arch Intern Med 163:18321836.Chrousos GP (1995) The hypothalamicpituitaryadrenal axis and immunemediated inflammation. N Engl J Med 332:13511362.Church MK, elLati S and Caulfield JP (1991) Neuropeptideinduced secretion from human skin mast cells. Int Arch Allergy Appl Immunol 94:310318.Cleare AJ, Miell J, Heap E, Sookdeo S, Young L, Malhi GS and O'Keane V (2001)Hypothalamopituitaryadrenal axis dysfunction in chronic fatigue syndrome, and the effects of lowdose hydrocortisone therapy. J Clin Endocrinol Metab 86:35453554.Collatz A, Johnston SC, Staines DR and MarshallGradisnik SM (2016) A Systematic Review of Drug Therapies for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Clin Ther 38:12631271.Collin SM, Bakken IJ, Nazareth I, Crawley E and White PD (2017) Trends in the incidence of chronic fatigue syndrome and fibromyalgia in the UK, 20012013: a Clinical Practice Research Datalink study. J R Soc Med 110:231244.Collins LV, Hajizadeh S, Holme E, Jonsson IM and Tarkowski A (2004) Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 75:9951000.Conti P, Caraffa A, Kritas SK, Ronconi G, Lessiani G, Toniato E and Theoharides TC (2017) Mast cell, proinflammatory and antiinflammatory: Jekyll and Hyde, the story continues. J Biol Regul Homeost Agents 31:263267.Cook DB, Nagelkirk PR, Poluri A, Mores J and Natelson BH (2006) The influence of aerobic fitness and fibromyalgia on cardiorespiratory and perceptual responses to exercise in patients with chronic fatigue syndrome. Arthritis Rheum 54:33513362.Crawley E (2014) The epidemiology of chronic fatigue syndrome/myalgic encephalitis in children. Arch Dis Child 99:171174.Crawley E, Hunt L and Stallard P (2009) Anxiety in children with CFS/ME. Eur Child Adolesc Psychiatry 18:683689.Crompton R, Clifton VL, Bisits AT, Read MA, Smith R and Wright IM (2003) Corticotropinreleasing hormone causes vasodilation in human skin via mast celldependent pathways. J Clin Endocrinol Metab 88:54275432.Davis JM, Murphy EA, Carmichael MD and Davis B (2009) Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Physiol Regul Integr Comp Physiol 296:R1071R1077.Davis SD, Kator SF, Wonnett JA, Pappas BL and Sall JL (2000) Neurally mediated hypotension in fatigued Gulf War veterans: a preliminary report. Am J Med Sci 319:8995.Daviss B. (2005) Growing pains for metabolomics. The Scientist 19:2528.de Cavanagh EM, Inserra F and Ferder L (2011) Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res 89:3140.Dekker JM, Girman C, Rhodes T, Nijpels G, Stehouwer CD, Bouter LM and Heine RJ (2005) Metabolic syndrome and 10year cardiovascular disease risk in the Hoorn Study. Circulation 112:666673.Deqiu Z, Kang L, Jiali Y, Baolin L and Gaolin L (2011) Luteolin inhibits inflammatory response and improves insulin sensitivity in the endothelium. Biochimie 93:506512.Dietert RR and Dietert JM (2008) Possible role for earlylife immune insult including developmental immunotoxicity in chronic fatigue syndrome (CFS) or myalgic encephalomyelitis (ME). Toxicology 247:6172.Dinarello CA, NoldPetry C, Nold M, Fujita M, Li S, Kim S and Bufler P (2016) Suppression of innate inflammation and immunity by interleukin37. Eur J Immunol 46:10671081.Ding W and Liu Y (2011) Genistein attenuates genioglossus muscle fatigue under chronic intermittent hypoxia by downregulation of oxidative stress level and upregulation of antioxidant enzyme activity through ERK1/2 signaling pathway. Oral Dis 17:677684.Dominguez LJ, Barbagallo M, Lauretani F, Bandinelli S, Bos A, Corsi AM, Simonsick EM and Ferrucci L (2006) Magnesium and muscle performance in older persons: the InCHIANTI study. Am J Clin Nutr 84:419426.Donelan J, Boucher W, Papadopoulou N, Lytinas M, Papaliodis D and Theoharides TC (2006) Corticotropinreleasing hormone induces skin vascular permeability through a neurotensindependent process. Proc Natl Acad Sci USA 103:77597764.Douglas SD and Leeman SE (2011) Neurokinin1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Ann N Y Acad Sci 1217:8395.Edvinsson L, CervosNavarro J, Larsson LI, Owman C and Ronnberg AL (1977) Regional distribution of mast cells containing histamine, dopamine or 5hydroxytryptamine in the mammalian brain. Neurology 27:878884.Esposito P, Chandler N, KandereGrzybowska K, Basu S, Jacobson S, Connolly R, Tutor D and Theoharides TC (2002) Corticotropinreleasing hormone (CRH) and brain mast cells regulate bloodbrainbarrier permeability induced by acute stress. J Pharmacol Exp Ther 303:10611066.Filler K, Lyon D, Bennett J, McCain N, Elswick R, Lukkahatai N and Saligan LN (2014) Association of Mitochondrial Dysfunction and Fatigue: A Review of the Literature. BBA Clin 1:1223.Fletcher MA, Rosenthal M, Antoni M, Ironson G, Zeng XR, Barnes Z, Harvey JM, Hurwitz B, Levis S, Broderick G and Klimas NG (2010) Plasma neuropeptide Y: a biomarker for symptom severity in chronic fatigue syndrome. Behav Brain Funct 6:76.Fluge O, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, Rekeland IG, Sapkota D, RoslandGV, Fossa A, KtoridouValen I, Lunde S, Sorland K, Lien K, Herder I, Thurmer H, Gotaas ME,Baranowska KA, Bohnen LM, Schafer C, McCann A, Sommerfelt K, Helgeland L, Ueland PM, Dahl O and Tronstad KJ (2016) Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 1:e89376.Fremont M, Metzger K, Rady H, Hulstaert J and De MK (2009) Detection of herpesviruses and parvovirus B19 in gastric and intestinal mucosa of chronic fatigue syndrome patients. In vivo 23:209213.Frith J, Zalewski P, Klawe JJ, Pairman J, Bitner A, TafilKlawe M and Newton JL (2012) Impaired blood pressure variability in chronic fatigue syndrome\a potential biomarker. QJM 105:831838.Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG and Komaroff A (1994) The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med 121:953959.Galli SJ, Tsai M and Piliponsky AM (2008) The development of allergic inflammation. Nature 454:445454.Ganiats TG (2015) Redefining the chronic fatigue syndrome. Ann Intern Med 162:653654.Georgiades E, Behan WM, Kilduff LP, Hadjicharalambous M, Mackie EE, Wilson J, Ward SA and Pitsiladis YP (2003) Chronic fatigue syndrome: new evidence for a central fatigue disorder. Clin Sci (Lond) 105:213218.Germain A, Ruppert D, LeVine SM and Hanson MR (2017) Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol Biosyst 13:371379.Gerwyn M and Maes M (2017) Mechanisms Explaining Muscle Fatigue and Muscle Pain in Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): a Review of Recent Findings. Curr Rheumatol Rep 19:1.Glassford JA (2017) The Neuroinflammatory Etiopathology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front Physiol 8:88.Gong J, Yang NS, Croft M, Weng IC, Sun L, Liu FT and Chen SS (2010) The antigen presentation function of bone marrowderived mast cells is spatiotemporally restricted to a subset expressing high levels of cell surface FcepsilonRI and MHC II. BMC Immunol 11:34.Gorman GS, Elson JL, Newman J, Payne B, McFarland R, Newton JL and Turnbull DM (2015) Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul Disord 25:563566.Guo W, Kong E and Meydani M (2009) Dietary polyphenols, inflammation, and cancer. Nutr Cancer 61:807810.Gupta A, Vij G and Chopra K (2010) Possible role of oxidative stress and immunological activation in mouse model of chronic fatigue syndrome and its attenuation by olive extract. J Neuroimmunol 226:37.Gupta A, Vij G, Sharma S, Tirkey N, Rishi P and Chopra K (2009) Curcumin, a polyphenolic antioxidant, attenuates chronic fatigue syndrome in murine water immersion stress model. Immunobiology 214:3339.Gwini SM, Forbes AB, Sim MR and Kelsall HL (2016) Multisymptom Illness in Gulf War Veterans: A Systematic Review and MetaAnalysis. J Occup Environ Med 58:659667.Haney E, Smith ME, McDonagh M, Pappas M, Daeges M, Wasson N and Nelson HD (2015)Diagnostic Methods for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Systematic Review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med 162:834840.Hanson SJ, Gause W and Natelson B (2001) Detection of immunologically significant factors for chronic fatigue syndrome using neuralnetwork classifiers. Clin Diagn Lab Immunol 8:658662.Harwood M, nielewskaNikiel B, Borzelleca JF, Flamm GW, Williams GM and Lines TC (2007) A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem Toxicol 45:21792205.Hokfelt T, Pernow B and Wahren J (2001) Substance P: a pioneer amongst neuropeptides. J Intern Med 249:2740.Hollingsworth KG, Jones DE, Taylor R, Blamire AM and Newton JL (2010) Impaired cardiovascular response to standing in chronic fatigue syndrome. Eur J Clin Invest 40:608615.Hornig M, Gottschalk CG, Eddy ML, Che X, Ukaigwe JE, Peterson DL and Lipkin WI (2017) Immune network analysis of cerebrospinal fluid in myalgic encephalomyelitis/chronic fatigue syndrome with atypical and classical presentations. Transl Psychiatry 7:e1080.Hornig M, Gottschalk G, Peterson DL, Knox KK, Schultz AF, Eddy ML, Che X and Lipkin WI (2016) Cytokine network analysis of cerebrospinal fluid in myalgic encephalomyelitis/chronic fatigue syndrome. Mol Psychiatry 21:261269.Hornig M, Montoya JG, Klimas NG, Levine S, Felsenstein D, Bateman L, Peterson DL and Gottschalk LR (2015) Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci Adv 1:e1400121.Ishisaka A, Ichikawa S, Sakakibara H, Piskula MK, Nakamura T, Kato Y, Ito M, Miyamoto K, Tsuji A, Kawai Y and Terao J (2011) Accumulation of orally administered quercetin in brain tissue and its antioxidative effects in rats. Free Radic Biol Med 51:13291336.Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, Taskinen MR and Groop L (2001) Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care 24:683689.Jang S, Kelley KW and Johnson RW (2008) Luteolin reduces IL6 production in microglia by inhibiting JNK phosphorylation and activation of AP1. Proc Natl Acad Sci USA 105:75347539.Jason LA, Benton MC, Valentine L, Johnson A and TorresHarding S (2008) The economic impact of ME/CFS: individual and societal costs. Dyn Med 7:6.Jason LA, Jordan K, Miike T, Bell DS, Lapp C, TorresHarding S, Rowe K, Gurwitt A, De Meirleir K, Van Hoof ELS and Psych C (2006) A pediatric case definition for myalgic encephalomyelitis and chronic fatigue syndrome. Journal of Chronic Fatigue Syndrome 13:144.Jason LA, Porter N, Brown M, Anderson V, Brown A, Hunnell J and Lerch A (2009) CFS: A Review of Epidemiology and Natural History Studies. Bull IACFS ME 17:88106.Jiang W, Babyak M, Krantz DS, Waugh RA, Coleman E, Hanson MM, Frid DJ, McNulty S, Morris JJ, O'Connor CM and Blumenthal JA (1996) Mental stressinduced myocardial ischemia and cardiac events. JAMA 275:16511656.Johri A, Calingasan NY, Hennessey TM, Sharma A, Yang L, Wille E, Chandra A and Beal MF (2012) Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington's disease. Hum Mol Genet 21:11241137.Jones MG, Cooper E, Amjad S, Goodwin CS, Barron JL and Chalmers RA (2005) Urinary and plasma organic acids and amino acids in chronic fatigue syndrome. Clin Chim Acta 361:150158.Kass LS and Poeira F (2015) The effect of acute vs chronic magnesium supplementation on exercise and recovery on resistance exercise, blood pressure and total peripheral resistance on normotensive adults. J Int Soc Sports Nutr 12:19.Katz BZ, Shiraishi Y, Mears CJ, Binns HJ and Taylor R (2009) Chronic fatigue syndrome after infectious mononucleosis in adolescents. Pediatrics 124:189193.Kaur J (2014) A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014:943162.Kawanishi S, Oikawa S and Murata M (2005) Evaluation for safety of antioxidant chemopreventive agents. Antioxid Redox Signal 7:17281739.Keller BA, Pryor JL and Giloteaux L (2014) Inability of myalgic encephalomyelitis/chronic fatigue syndrome patients to reproduce VO(2)peak indicates functional impairment. J Transl Med 12:104.Kempuraj D, Madhappan B, Christodoulou S, Boucher W, Cao J, Papadopoulou N, Cetrulo CL and Theoharides TC (2005) Flavonols inhibit proinflammatory mediator release, intracellular calcium ion levels and protein kinase C theta phosphorylation in human mast cells. Br J Pharmacol 145:934944.Kempuraj D, Papadopoulou NG, Lytinas M, Huang M, KandereGrzybowska K, Madhappan B, Boucher W, Christodoulou S, Athanassiou A and Theoharides TC (2004) Corticotropinreleasing hormone and its structurally related urocortin are synthesized and secreted by human mast cells. Endocrinology 145:4348.Kennedy G, Spence VA, McLaren M, Hill A, Underwood C and Belch JJ (2005) Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med 39:584589.Klimas NG, Broderick G and Fletcher MA (2012) Biomarkers for chronic fatigue. Brain Behav Immun 26:12021210.Komaroff AL (2015) Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Real Illness. Ann Intern Med 162:871872.Krantz DS, Sheps DS, Carney RM and Natelson BH (2000) Effects of mental stress in patients with coronary artery disease: evidence and clinical implications. JAMA 283:18001802.Kuo YH, Tsai WJ, Loke SH, Wu TS and Chiou WF (2009) Astragalus membranaceus flavonoids (AMF) ameliorate chronic fatigue syndrome induced by food intake restriction plus forced swimming. J Ethnopharmacol 122:2834.Kurokawa H, Sugiyama S, Nozaki T, Sugamura K, Toyama K, Matsubara J, Fujisue K, Ohba K,Maeda H, Konishi M, Akiyama E, Sumida H, Izumiya Y, Yasuda O, KimMitsuyama S and Ogawa H (2015) Telmisartan enhances mitochondrial activity and alters cellular functions in human coronary artery endothelial cells via AMPactivated protein kinase pathway. Atherosclerosis 239:375385.Leeman SE and Carraway RE (1982) Neurotensin: discovery, isolation, characterization, synthesis and possible physiological roles. Ann N Y Acad Sci 400:116.Li C, Ford ES, McGuire LC and Mokdad AH (2007) Association of metabolic syndrome and insulin resistance with congestive heart failure: findings from the Third National Health and Nutrition Examination Survey. J Epidemiol Community Health 61:6773.Libby P, Ridker PM and Maseri A (2002) Inflammation and atherosclerosis. Circulation 105:11351143.Liew FY, Pitman NI and McInnes IB (2010) Diseaseassociated functions of IL33: the new kid in the IL1 family. Nat Rev Immunol 10:103110.Loades ME, Sheils EA and Crawley E (2016) Treatment for paediatric chronic fatigue syndrome or myalgic encephalomyelitis (CFS/ME) and comorbid depression: a systematic review. BMJ Open 6:e012271.Loevinger BL, Muller D, Alonso C and Coe CL (2007) Metabolic syndrome in women with chronic pain. Metabolism 56:8793.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH and Kipnis J (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337341.Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N and Bosmans E (2009) Coenzyme Q10 deficiency in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is related to fatigue, autonomic and neurocognitive symptoms and is another risk factor explaining the early mortality in ME/CFS due to cardiovascular disorder. Neuro Endocrinol Lett 30:470476.Maes M, Twisk FN and Johnson C (2012) Myalgic Encephalomyelitis (ME), Chronic Fatigue Syndrome (CFS), and Chronic Fatigue (CF) are distinguished accurately: results of supervised learning techniques applied on clinical and inflammatory data. Psychiatry Res 200:754760.Maes M, Twisk FN, Kubera M and Ringel K (2011) Evidence for inflammation and activation of cellmediated immunity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Increased interleukin1, tumor necrosis factoralpha, PMNelastase, lysozyme and neopterin. J Affect Disord 136:933939.Maloney EM, Boneva R, Nater UM and Reeves WC (2009) Chronic fatigue syndrome and high allostatic load: results from a populationbased casecontrol study in Georgia. Psychosom Med 71:549556.Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, Lopes GA, Russo RC,Avila TV, Melgaco JG, Oliveira AG, Pinto MA, Lima CX, De Paula AM, Cara DC, Leite MF, Teixeira MM and Menezes GB (2012) Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56:19711982.MartinezMartinez LA, Mora T, Vargas A, FuentesIniestra M and MartinezLavin M (2014) Sympathetic nervous system dysfunction in fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome, and interstitial cystitis: a review of casecontrol studies. J Clin Rheumatol 20:146150.Mashaghi A, Marmalidou A, Tehrani M, Grace PM, Pothoulakis C and Dana R (2016) Neuropeptide substance P and the immune response. Cell Mol Life Sci 73:42494264.Mastrangelo F, Frydas I, Ronconi G, Kritas SK, Tettamanti L, Caraffa A, Ovidio D, Younes A, Gallenga CE and Conti P (2018) Lowgrade chronic inflammation mediated by mast cells in fibromyalgia: role of IL37. J Biol Regul Homeost Agents 32:195198.Matusik P, Guzik B, Weber C and Guzik TJ (2012) Do we know enough about the immune pathogenesis of acute coronary syndromes to improve clinical practice? Thromb Haemost 108:443456.McCully KK and Natelson BH (1999) Impaired oxygen delivery to muscle in chronic fatigue syndrome. Clin Sci (Lond) 97:603608.McCully KK, Smith S, Rajaei S, Leigh JS, Jr. and Natelson BH (2004) Muscle metabolism with blood flow restriction in chronic fatigue syndrome. J Appl Physiol (1985 ) 96:871878.Meeus M, Goubert D, De BF, Struyf F, Hermans L, Coppieters I, De W, I, Da SH and Calders P (2013) Heart rate variability in patients with fibromyalgia and patients with chronic fatigue syndrome: a systematic review. Semin Arthritis Rheum 43:279287.Middleton EJ, Kandaswami C and Theoharides TC (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease and cancer. Pharmacol Rev 52:673751.Mitsuishi M, Miyashita K, Muraki A and Itoh H (2009) Angiotensin II reduces mitochondrial content in skeletal muscle and affects glycemic control. Diabetes 58:710717.Miwa K and Fujita M (2009) Cardiovascular dysfunction with low cardiac output due to a small heart in patients with chronic fatigue syndrome. Intern Med 48:18491854.Monro JA and Puri BK (2018) A Molecular Neurobiological Approach to Understanding the Aetiology of Chronic Fatigue Syndrome (Myalgic Encephalomyelitis or Systemic Exertion Intolerance Disease) with Treatment Implications. Mol Neurobiol.Morris G, Anderson G and Maes M (2016) HypothalamicPituitaryAdrenal Hypofunction inMyalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated ImmuneInflammatory and Oxidative and Nitrosative Pathways. Mol Neurobiol 54:68066819.Morris G, Berk M, Galecki P and Maes M (2014) The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol Neurobiol 49:741756.Morris G and Maes M (2012) Increased nuclear factorkappaB and loss of p53 are key mechanisms in Myalgic Encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Hypotheses 79:607613.Morris G and Maes M (2013) Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med 11:205.Morris G and Maes M (2014) Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immunoinflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis 29:1936.Morris G, Stubbs B, Kohler CA, Walder K, Slyepchenko A, Berk M and Carvalho AF (2018) The putative role of oxidative stress and inflammation in the pathophysiology of sleep dysfunction across neuropsychiatric disorders: Focus on chronic fatigue syndrome, bipolar disorder and multiple sclerosis. Sleep Med Rev.Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL and Eisenberg MJ (2010) The metabolic syndrome and cardiovascular risk a systematic review and metaanalysis. J Am Coll Cardiol 56:11131132.Moulin D, Donze O, TalabotAyer D, Mezin F, Palmer G and Gabay C (2007) Interleukin (IL)33 induces the release of proinflammatory mediators by mast cells. Cytokine 40:216225.Mousli M and Landry Y (1994) Role of positive charges of neuropeptide Y fragments in mast cell activation. Agents Actions 41 Spec No:C41C42.Mukai K, Tsai M, Saito H and Galli SJ (2018) Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 282:121150.Murrough JW, Mao X, Collins KA, Kelly C, Andrade G, Nestadt P, LeVine SM, Mathew SJ and Shungu DC (2010) Increased ventricular lactate in chronic fatigue syndrome measured by 1H MRS imaging at 3.0 T. II: comparison with major depressive disorder. NMR Biomed 23:643650.Mustain WC, Rychahou PG and Evers BM (2011) The role of neurotensin in physiologic and pathologic processes. Curr Opin Endocrinol Diabetes Obes 18:7582.Myhill S, Booth NE and LarenHoward J (2009) Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med 2:116.Myhill S, Booth NE and LarenHoward J (2013) Targeting mitochondrial dysfunction in the treatment of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) a clinical audit. Int J Clin Exp Med 6:115.Nacul LC, Lacerda EM, Pheby D, Campion P, Molokhia M, Fayyaz S, Leite JC, Poland F, HoweA and Drachler ML (2011) Prevalence of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) in three regions of England: a repeated crosssectional study in primary care. BMC Med 9:91.Nadjar A, Wigren HM and Tremblay ME (2017) Roles of Microglial Phagocytosis andInflammatory Mediators in the Pathophysiology of Sleep Disorders. Front Cell Neurosci 11:250.NagySzakal D, Barupal DK, Lee B, Che X, Williams BL, Kahn EJR, Ukaigwe JE, Bateman L, Klimas NG, Komaroff AL, Levine S, Montoya JG, Peterson DL, Levin B, Hornig M, Fiehn O and Lipkin WI (2018) Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Sci Rep 8:10056.Najam SS, Zglinicki B, Vinnikov IA and Konopka W (2018) MicroRNAs in the hypothalamic control of energy homeostasis. Cell Tissue Res.Nakamura T, Schwander S, Donnelly R, Cook DB, Ortega F, Togo F, Yamamoto Y, Cherniack NS, Klapholz M, Rapoport D and Natelson BH (2013) Cytokines do not change after exercise or sleep deprivation in chronic fatigue syndrome. Clin Vaccine Immunol.Nakamura T, Schwander SK, Donnelly R, Ortega F, Togo F, Broderick G, Yamamoto Y, Cherniack NS, Rapoport D and Natelson BH (2010) Cytokines across the night in chronic fatigue syndrome with and without fibromyalgia. Clin Vaccine Immunol 17:582587.Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, Onoe K, Fukuda S, Kawabe J,Takahashi K, Kataoka Y, Shiomi S, Yamaguti K, Inaba M, Kuratsune H and Watanabe Y (2014) Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An 11C\(R)\PK11195 PET Study. J Nucl Med 55:945950.Naschitz J, Dreyfuss D, Yeshurun D and Rosner I (2004) Midodrine treatment for chronic fatigue syndrome. Postgrad Med J 80:230232.Natelson BH (2001) Chronic fatigue syndrome. JAMA 285:25572559.Natelson BH, Intriligator R, Cherniack NS, Chandler HK and Stewart JM (2007) Hypocapnia is a biological marker for orthostatic intolerance in some patients with chronic fatigue syndrome. Dyn Med 6:2.Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, Baxter A, Nathan N, Anderson W and Gordon E (2016) Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci U S A 113:E5472E5480.Newhouse IJ and Finstad EW (2000) The effects of magnesium supplementation on exercise performance. Clin J Sport Med 10:195200.Newton JL, Sheth A, Shin J, Pairman J, Wilton K, Burt JA and Jones DE (2009) Lower ambulatory blood pressure in chronic fatigue syndrome. Psychosom Med 71:361365.Nguyen T, Johnston S, Chacko A, Gibson D, Cepon J, Smith P, Staines D and MarshallGradisnik S (2017) Novel characterisation of mast cell phenotypes from peripheral blood mononuclear cells in chronic fatigue syndrome/myalgic encephalomyelitis patients. Asian Pac J Allergy Immunol 35:7581.Niblett SH, King KE, Dunstan RH, CliftonBligh P, Hoskin LA, Roberts TK, Fulcher GR, McGregor NR, Dunsmore JC, Butt HL, Klineberg I and Rothkirch TB (2007) Hematologic and urinary excretion anomalies in patients with chronic fatigue syndrome. Exp Biol Med (Maywood ) 232:10411049.Nijhof SL, Maijer K, Bleijenberg G, Uiterwaal CS, Kimpen JL and van de Putte EM (2011) Adolescent chronic fatigue syndrome: prevalence, incidence, and morbidity. Pediatrics 127:e1169e1175.Nijs J, Nees A, Paul L, De KM, Ickmans K, Meeus M and Van OJ (2014) Altered immune response to exercise in patients with chronic fatigue syndrome/myalgic encephalomyelitis: a systematic literature review. Exerc Immunol Rev 20:94116.O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP and Shanahan F (2004) The role of substance P in inflammatory disease. J Cell Physiol 201:167180.O'Kane M, Murphy EP and Kirby B (2006) The role of corticotropinreleasing hormone in immunemediated cutaneous inflammatory disease. Exp Dermatol 15:143153.Ozturan A, Eyigor H, Eyigor M, Osma U, Yilmaz MD, Selcuk OT, Renda L and Gultekin M (2017) The role of IL25 and IL33 in chronic rhinosinusitis with or without nasal polyps. Eur Arch Otorhinolaryngol 274:283288.Pae CU, Marks DM, Patkar AA, Masand PS, Luyten P and Serretti A (2009) Pharmacological treatment of chronic fatigue syndrome: focusing on the role of antidepressants. Expert Opin Pharmacother 10:15611570.Pang X, Letourneau R, Rozniecki JJ, Wang L and Theoharides TC (1996) Definitive characterization of rat hypothalamic mast cells. Neuroscience 73:889902.Patel AB and Theoharides TC (2017) Methoxyluteolin Inhibits Neuropeptidestimulated Proinflammatory Mediator Release via mTOR Activation from Human Mast Cells. J Pharmacol Exp Ther 361:462471.Patel AB, Tsilioni I, Leeman SE and Theoharides TC (2016) Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc Natl Acad Sci U S A 113:E7049E7058.Pejovic S, Natelson BH, Basta M, FernandezMendoza J, Mahr F and Vgontzas AN (2015) Chronic fatigue syndrome and fibromyalgia in diagnosed sleep disorders: a further test of the 'unitary' hypothesis. BMC Neurol 15:53.Petersen KF and Shulman GI (2006) Etiology of insulin resistance. Am J Med 119:S10S16.Peterson PK, Pheley A, Schroeppel J, Schenck C, Marshall P, Kind A, Haugland JM, Lambrecht LJ, Swan S and Goldsmith S (1998) A preliminary placebocontrolled crossover trial of fludrocortisone for chronic fatigue syndrome. Arch Intern Med 158:908914.Petra AI, Panagiotidou S, Hatziagelaki E, Stewart JM, Conti P and Theoharides TC (2015) GutMicrobiotaBrain Axis and Its Effect on Neuropsychiatric Disorders With Suspected Immune Dysregulation. Clin Ther 37:984995.Petty RD, McCarthy NE, Le DR and Kerr JR (2016) MicroRNAs hsamiR99b, hsamiR330, hsamiR126 and hsamiR30c: Potential Diagnostic Biomarkers in Natural Killer (NK) Cells of Patients with Chronic Fatigue Syndrome (CFS)/ Myalgic Encephalomyelitis (ME). PLoS ONE 11:e0150904.Pranzatelli MR (2018) Advances in BiomarkerGuided Therapy for Pediatric\ and AdultOnset Neuroinflammatory Disorders: Targeting Chemokines/Cytokines. Front Immunol 9:557.Ravindran MK, Zheng Y, Timbol C, Merck SJ and Baraniuk JN (2011) Migraine headaches in chronic fatigue syndrome (CFS): comparison of two prospective crosssectional studies. BMC Neurol 11:30.RepkaRamirez S, Naranch K, Park YJ, Clauw D and Baraniuk JN (2002) Cytokines in nasal lavage fluids from acute sinusitis, allergic rhinitis, and chronic fatigue syndrome subjects. Allergy Asthma Proc 23:185190.Robuffo I, Toniato E, Tettamanti L, Mastrangelo F, Ronconi G, Frydas I, Caraffa A, Kritas SK and Conti P (2017) Mast cell in innate immunity mediated by proinflammatory and antiinflammatory IL1 family members. J Biol Regul Homeost Agents 31:837842.Rowe PC, Fontaine KR, Lauver M, Jasion SE, Marden CL, Moni M, Thompson CB and Violand RL (2016) Neuromuscular Strain Increases Symptom Intensity in Chronic Fatigue Syndrome. PLoS ONE 11:e0159386.Sachdeva AK, Kuhad A and Chopra K (2011) Epigallocatechin gallate ameliorates behavioral and biochemical deficits in rat model of loadinduced chronic fatigue syndrome. Brain Res Bull 86:165172.Sachdeva AK, Kuhad A, Tiwari V and Chopra K (2009) Epigallocatechin gallate ameliorates chronic fatigue syndrome in mice: Behavioral and biochemical evidence. Behav Brain Res 205:414420.Saluja R, Ketelaar ME, Hawro T, Church MK, Maurer M and Nawijn MC (2015) The role of the IL33/IL1RL1 axis in mast cell and basophil activation in allergic disorders. Mol Immunol 63:8085.Scheibenbogen C, Freitag H, Blanco J, Capelli E, Lacerda E, Authier J, Meeus M, Castro MJ,NoraKrukle Z, Oltra E, Strand EB, Shikova E, Sekulic S and Murovska M (2017) The European ME/CFS Biomarker Landscape project: an initiative of the European network EUROMENE. J Transl Med 15:162.Schoeman EM, Van Der Westhuizen FH, Erasmus E, van DE, Knowles CV, AlAli S, Ng WF, Taylor RW, Newton JL and Elson JL (2017) Clinically proven mtDNA mutations are not common in those with chronic fatigue syndrome. BMC Med Genet 18:29.Shungu DC, Weiduschat N, Murrough JW, Mao X, Pillemer S, Dyke JP, Medow MS, Natelson BH, Stewart JM and Mathew SJ (2012a) Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR Biomed 25:10731087.Shungu DC, Weiduschat N, Murrough JW, Mao X, Pillemer S, Dyke JP, Medow MS, Natelson BH, Stewart JM and Mathew SJ (2012b) Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR Biomed 25:10731087.Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M and Chaudhary D (2010) IL33 synergizes with IgEdependent and IgEindependent agents to promote mast cell and basophil activation. Inflamm Res 59:207218.Sismanopoulos N, Delivanis DA, Mavrommati D, Hatziagelaki E, Conti P and Theoharides TC (2013) Do mast cells link obesity and asthma? Allergy 68:815.Slominski A (2009) On the role of the corticotropinreleasing hormone signalling system in the aetiology of inflammatory skin disorders. Br J Dermatol 160:229232.Smith MS, MartinHerz SP, Womack WM and Marsigan JL (2003) Comparative study of anxiety, depression, somatization, functional disability, and illness attribution in adolescents with chronic fatigue or migraine. Pediatrics 111:e376e381.Smits B, van den HL, Knoop H, Kusters B, Janssen A, Borm G, Bleijenberg G, Rodenburg R and van EB (2011) Mitochondrial enzymes discriminate between mitochondrial disorders and chronic fatigue syndrome. Mitochondrion 11:735738.Sotzny F, Blanco J, Capelli E, CastroMarrero J, Steiner S, Murovska M and Scheibenbogen C (2018) Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Evidence for an autoimmune disease. Autoimmun Rev 17:601609.Suarez A, Guillamo E, Roig T, Blazquez A, Alegre J, Bermudez J, Ventura JL, GarciaQuintana AM, Comella A, Segura R and Javierre C (2010) Nitric oxide metabolite production during exercise in chronic fatigue syndrome: a casecontrol study. J Womens Health (Larchmt ) 19:10731077.Sumbalova Z, Kucharska J and Kristek F (2010) Losartan improved respiratory function and coenzyme Q content in brain mitochondria of young spontaneously hypertensive rats. Cell Mol Neurobiol 30:751758.Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D and Zhang HG (2010) A novel nanoparticle drug delivery system: the antiinflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther 18:16061614.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ and Itagaki K (2013) Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PloS One 8:e59989.Swindle EJ and Metcalfe DD (2007) The role of reactive oxygen species and nitric oxide in mast celldependent inflammatory processes. Immunol Rev 217:186205.Taildeman J, PerezNovo CA, Rottiers I, Ferdinande L, Waeytens A, De C, V, Bachert C, Demetter P, Waelput W, Braet K and Cuvelier CA (2009) Human mast cells express leptin and leptin receptors. Histochem Cell Biol 131:703711.Takeuchi K, Yamamoto K, Ohishi M, Takeshita H, Hongyo K, Kawai T, Takeda M, Kamide K, Kurtz TW and Rakugi H (2013) Telmisartan modulates mitochondrial function in vascular smooth muscle cells. Hypertens Res 36:433439.Taliou A, Zintzaras E, Lykouras L and Francis K (2013) An openlabel pilot study of a formulation containing the antiinflammatory flavonoid luteolin and its effects on behavior in children with autism spectrum disorders. Clin Ther 35:592602.Taracanova A, Alevizos M, Karagkouni A, Weng Z, Norwitz E, Conti P, Leeman SE and Theoharides TC (2017c) SP and IL33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc Natl Acad Sci U S A 114:E4002E4009.Taracanova A, Alevizos M, Karagkouni A, Weng Z, Norwitz E, Conti P, Leeman SE andTheoharides TC (2017a) SP and IL33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc Natl Acad Sci U S A 114:E4002E4009.Taracanova A, Alevizos M, Karagkouni A, Weng Z, Norwitz E, Conti P, Leeman SE andTheoharides TC (2017b) SP and IL33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc Natl Acad Sci U S A 114:E4002E4009.Theoharides TC., Tsilioni I, Patel AB and Doyle R. (2016) Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry 6:e844.Theoharides TC (1990) Mast cells: the immune gate to the brain. Life Sci 46:607617.Theoharides TC (2013) Atopic conditions in search of pathogenesis and therapy. Clin Ther 35:544547.Theoharides TC (2016) Danger Signals and Inflammation. Clin Ther 38:996999.Theoharides TC (2017) Neuroendocrinology of mast cells: Challenges and controversies. Exp Dermatol 26:751759.Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A and Kalogeromitros D (2010a) Mast cells and inflammation. Biochim Biophys Acta 1822:2133.Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A and Kalogeromitros D (2010c) Mast cells and inflammation. Biochim Biophys Acta 1822:2133.Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A and Kalogeromitros D (2010b) Mast cells and inflammation. Biochim Biophys Acta 1822:2133.Theoharides TC, Asadi S, Panagiotidou S and Weng Z (2013) The "missing link" in autoimmunity and autism: Extracellular mitochondrial components secreted from activated live mast cells. Autoimmun Rev 12:11361142.Theoharides TC, Betchaku T and Douglas WW (1990) Somatostatininduced histamine secretion in mast cells; a comparison with known mast cell secretagogues. J Biol Chem.Theoharides TC and Cochrane DE (2004) Critical role of mast cells in inflammatory diseases and the effect of acute stress. J Neuroimmunol 146:112.Theoharides TC, Conti P and Economu M (2014) Brain inflammation, neuropsychiatric disorders, and immunoendocrine effects of luteolin. J Clin Psychopharmacol 34:187189.Theoharides TC, Donelan JM, Papadopoulou N, Cao J, Kempuraj D and Conti P (2004a) Mast cells as targets of corticotropinreleasing factor and related peptides. Trends Pharmacol Sci 25:563568.Theoharides TC and Kalogeromitros D (2006) The critical role of mast cells in allergy and inflammation. Ann N Y Acad Sci 1088:7899.Theoharides TC, Kempuraj D, Tagen M, Conti P and Kalogeromitros D (2007) Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol Rev 217:6578.Theoharides TC and Konstantinidou A (2007) Corticotropinreleasing hormone and the bloodbrainbarrier. Front Biosci 12:16151628.Theoharides TC, Makris M and Kalogeromitros D (2008) Allergic inflammation and adipocytokines. Int J Immunopathol Pharmacol 21:14.Theoharides TC, Petra AI, Taracanova A, Panagiotidou S and Conti P (2015a) Targeting IL33 in autoimmunity and inflammation. J Pharmacol Exp Ther 354:2431.Theoharides TC, Singh LK, Boucher W, Pang X, Letourneau R, Webster E and Chrousos G (1998) Corticotropinreleasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 139:403413.Theoharides TC, Sismanopoulos N, Delivanis DA, Zhang B, Hatziagelaki EE and Kalogeromitros D (2011) Mast cells squeeze the heart and stretch the gird: Their role in atherosclerosis and obesity. Trends Pharmacol Sci 32:534542.Theoharides TC and Stewart JM (2016) PostLyme SyndromeAssociated Polyneuropathy Treated With Immune Immunoglobulin and a LuteolinContaining Formulation. J Clin Psychopharmacol 36:290291.Theoharides TC, Stewart JM, Hatziagelaki E and Kolaitis G (2015b) Brain "fog," inflammation and obesity: key aspects of neuropsychiatric disorders improved by luteolin. Front Neurosci 9:225.Theoharides TC, Tsilioni I, Arbetman L, Panagiotidou S, Stewart JM, Gleason RM and Russell IJ (2015c) Fibromyalgia, a syndrome in search of pathogenesis and therapy. J Pharmacol Exp Ther 355:255263.Theoharides TC, Valent P and Akin C (2015d) Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med 373:163172.Theoharides TC, Weinkauf C and Conti P (2004b) Brain cytokines and neuropsychiatric disorders. J Clin Psychopharmacol 24:577581.Theoharides TC, Zhang B, Kempuraj D, Tagen M, Vasiadi M, Angelidou A, Alysandratos KD, Kalogeromitros D, Asadi S, Stavrianeas N, Peterson E, Leeman S and Conti P (2010d) IL33 augments substance Pinduced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci U S A 107:44484453.Theoharides TC, Zhang B, Kempuraj D, Tagen M, Vasiadi M, Angelidou A, Alysandratos KD, Kalogeromitros D, Asadi S, Stavrianeas N, Peterson E, Leeman S and Conti P (2010e) IL33 augments substance Pinduced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci U S A 107:44484453.Tomas C, Brown A, Strassheim V, Elson J, Newton J and Manning P (2017) Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 12:e0186802.Tomas C and Newton J (2018) Metabolic abnormalities in chronic fatigue syndrome/myalgic encephalomyelitis: a minireview. Biochem Soc Transpii: BST20170503.Toniato E, Frydas I, Robuffo I, Ronconi G, Caraffa A, Kritas SK and Conti P (2017) Activation and inhibition of adaptive immune response mediated by mast cells. J Biol Regul Homeost Agents 31:543548.Trivedi MS, Oltra E, Sarria L, Rose N, Beljanski V, Fletcher MA, Klimas NG and Nathanson L (2018) Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndromeassociated DNA methylation patterns. PLoS ONE 13:e0201066.Tsai AG, Wadden TA, Sarwer DB, Berkowitz RI, Womble LG, Hesson LA, Phelan S and Rothman R (2008) Metabolic syndrome and healthrelated quality of life in obese individuals seeking weight reduction. Obesity (Silver Spring) 16:5963.Tsilioni I, Taliou A, Francis K and Theoharides TC (2015) Children with Autism Spectrum Disorders, who improved with a luteolin containing dietary formulation, show reduced serum levels of TNF and IL6. Transl Psychiatry 5:e647.Tung HY, Plunkett B, Huang SK and Zhou Y (2014) Murine mast cells secrete and respond to interleukin33. J Interferon Cytokine Res 34:141147.Unger ER, Lin JS, Brimmer DJ, Lapp CW, Komaroff AL, Nath A, Laird S and Iskander J (2016) CDC Grand Rounds: Chronic Fatigue Syndrome Advancing Research and Clinical Education. MMWR Morb Mortal Wkly Rep 65:14341438.Valero T (2014) Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des 20:55075509.Vasiadi M, Newman J and Theoharides TC (2014) Isoflavones inhibit poly(I:C)\induced serum, brain, and skin inflammatory mediators relevance to chronic fatigue syndrome. J Neuroinflammation 11:168.Vermeulen RC, Kurk RM, Visser FC, Sluiter W and Scholte HR (2010) Patients with chronic fatigue syndrome performed worse than controls in a controlled repeated exercise study despite a normal oxidative phosphorylation capacity. J Transl Med 8:93.Vermeulen RC and Vermeulen vE, I (2014) Decreased oxygen extraction during cardiopulmonary exercise test in patients with chronic fatigue syndrome. J Transl Med 12:20.Vij G, Gupta A and Chopra K (2009) Modulation of antigeninduced chronic fatigue in mouse model of water immersion stress by naringin, a polyphenolic antioxidant. Fundam Clin Pharmacol 23:331337.Vincent A, Brimmer DJ, Whipple MO, Jones JF, Boneva R, Lahr BD, Maloney E, St Sauver JL and Reeves WC (2012) Prevalence, incidence, and classification of chronic fatigue syndrome in Olmsted County, Minnesota, as estimated using the Rochester Epidemiology Project. Mayo Clin Proc 87:11451152.Wang G, Pierangeli SS, Papalardo E, Ansari GA and Khan MF (2010) Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 62:20642072.Weng Z, Patel AB, Panagiotidou S and Theoharides TC (2015) The novel flavonetetramethoxyluteolin is a potent inhibitor of human mast cells. J Allergy Clin Immunol 135:10441052.Wernersson S and Pejler G (2014) Mast cell secretory granules: armed for battle. Nat Rev Immunol 14:478494.Whitmore KE and Theoharides TC (2011) When to suspect interstitial cystitis. J Fam Pract 60:340348.Wyller VB, Barbieri R and Saul JP (2011) Blood pressure variability and closedloop baroreflex assessment in adolescent chronic fatigue syndrome during supine rest and orthostatic stress. Eur J Appl Physiol 111:497507.Wyller VB, Saul JP, Walloe L and Thaulow E (2008) Sympathetic cardiovascular control during orthostatic stress and isometric exercise in adolescent chronic fatigue syndrome. Eur J Appl Physiol 102:623632.Xiao ZP, Peng ZY, Peng MJ, Yan WB, Ouyang YZ and Zhu HL (2011) Flavonoids health benefits and their molecular mechanism. Mini Rev Med Chem 11:169177.Xu H, Bin NR and Sugita S (2018) Diverse exocytic pathways for mast cell mediators. Biochem Soc Trans 46:235247.Xu M, Dai W and Deng X (2002) Effects of magnesium sulfate on brain mitochondrial respiratory function in rats after experimental traumatic brain injury. Chin J Traumatol 5:361364.Yamano E, Sugimoto M, Hirayama A, Kume S, Yamato M, Jin G, Tajima S, Goda N, Iwai K, Fukuda S, Yamaguti K, Kuratsune H, Soga T, Watanabe Y and Kataoka Y (2016) Index markers of chronic fatigue syndrome with dysfunction of TCA and urea cycles. Sci Rep 6:34990.Yancey JR and Thomas SM (2012) Chronic fatigue syndrome: diagnosis and treatment. Am Fam Physician 86:741746.Yang X, Zhu J and Tang Z (2007) Interventional effect of magnesium sulfate on nitric oxide synthase activity after acute craniocerebral injury. Neural Regen Res 2:251253.Yang TY, Kuo HT, Chen HJ, Chen CS, Lin WM, Tsai SY, Kuo CN and Kao CH (2015) Increased Risk of Chronic Fatigue Syndrome Following Atopy: A PopulationBased Study. Medicine (Baltimore) 94:e1211.Zhang B, Angelidou A, Alysandratos KD, Vasiadi M, Francis K, Asadi S, Theoharides A., Sideri k, Lykouras L, Kalogeromitros D and Theoharides TC (2010) Mitochondrial DNA and antimitochondrial antibodies in serum of autistic children. J Neuroinflammation 7:80.Zhang B, Alysandratos KD, Angelidou A, Asadi S, Sismanopoulos N, Delivanis DA, Weng Z,Miniati A, Vasiadi M, KatsarouKatsari A, Miao B, Leeman SE, Kalogeromitros D and Theoharides TC (2011) Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: Relevance to atopic dermatitis. J Allergy Clin Immunol 127:15221531.Zhang B, Asadi S, Weng Z, Sismanopoulos N and Theoharides TC (2012) Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PloS One 7:e49767.Zhang X, Wang Y, Dong H, Xu Y and Zhang S (2016) Induction of Microglial Activation by Mediators Released from Mast Cells. Cell Physiol Biochem 38:15201531.Zhang Y, Xun P, Wang R, Mao L and He K (2017) Can Magnesium Enhance Exercise Performance? Nutrients 9:pii: E946.
9) Hippocampus
10) HPA Axis
Much CFS research concerns the HPA axis, the images below show the relation between HPA axis and sleep
click for full size, click 'x' button to close
Conditions associated with fatigue, some more relevant than others
click here to download the page below
Page Synopsis: CFS and sleep disorder, If not for the PEM, POTS, IBS, Granuloma Annulare, drowsiness during wakefulness, and many other coconditions, CFS could be characterised as a sleep disorder
There is a theory (and evidence that CFS is a hibernation or 'Dauer' state (aka 'cell danger response' and that is addressed below
Hypersomnias (a class of sleep disorder is discussed as medicines for those may help CFS
This page has more detail and complexity than the others and is meant largely for research as it provides as many questions as answers
Skill Level 5
Relevance:3 Technical Level:5
relevance to the condition is a 5, however relevance to cures based on current medicine for CFS is 3
2) Cell Danger Response Theory
Cell Danger Response Theory
3) Sleep Disorders
page 32 CFS > ALLOPATHIC MEDICINES > SLEEP, SLEEP DISORDERS, APNEA MACHINE AND SNORE SURGERY
page 31
page 33
Page Table of contents
1) Overview
2) Fatigue (customary or chronic)
2a) Cognitive Fatigue
2b) Biological Processes Associated With Fatigue
2c) Diseases associated with fatigue (Disease Hierarchy)
2e) Fatigue (as Disease), Interaction with Substances
2f) Gene Interaction with Fatigue (as Disease)
2g) Molecular Functions Associated With Fatigue
2h) Pathways Associated With Fatigue
3) List of Sleep Disorders
International Classification of Sleep Disorders (ICSD), ICSD-3 Sleep Disorder Categories1. InsomniaThe ICSD-3 defines insomnia as "a repeated difficulty with sleep initiation, duration, consolidation, or quality that occurs despite adequate opportunity and circumstances for sleep, and results in some form of daytime impairment."The ICSD-3 groups insomnia into 4 major categories listed below.a. Chronic insomnia disorderb. Short-term insomnia disorderc. Other insomnia disorderd. Isolated symptoms and normal variant 2. Sleep-related breathing disordersThese disorders are divided into those of central origin (characterized by a lack of breathing effort) and those caused by an obstruction of the airways. a. Obstructive sleep apnea disordersi. Obstructive sleep apnea, adultii. Obstructive sleep apnea, pediatricb. Central sleep apnea syndromei. Central sleep apnea with Cheyne-Stokes breathingii. Central sleep apnea due to a medical disorder w/o Cheyne-Stokes breathingiii. Central sleep apnea due to high altitude periodic breathingiv. Central sleep apnea due to medicaiton or substancev. Primary central sleep apneavi. Primary central sleep apnea of infancyvii. Primary central sleep apnea of prematurityviii. Treatment-emergent central sleep apneac. Sleep-related hypoventilation disordersi. Obesity hypoventilation syndromeii. Congenital central alveoloar hypoventilation syndromeiii. Late-onset central hypoventilation with hypothalamic dysfunctioniv. Idiopathic central alveolar hypoventilationv. Sleep-related hypoventilation due to medication or substancevi. Sleep-related hypoventilation due to medical disorderd. Sleep-related hypoxemia disordere. Isolated symptoms and normal variantsi. Snoringii. Catathrenia 3. Central disorders of hypersomnolenceThe ICSD-3 categorizes this class of sleep disorders as those in which "the primary complaint is daytime sleepiness not caused by disturbed nocturnal sleep or misaligned circadian rhythms." a. Narcolepsy type Ib. Narcolepsy type IIc. Idiopathic hypersomniad. Kleine-Levin syndromee. Hypersomnia due to a medical disorderf. Hypersomnia due to a medication or substanceg. Hypersomnia associated with a psychiatric disorderh. Insufficient sleep syndrome 4. Circadian rhythm sleep-wake disordersThese disorders are characterized by a disturbance or disruption to the normal circadian rhythm, which causes patients to experience excessive daytime sleepiness, insomnia, or both.a. Delayed sleep-wake phase disorderb. Advanced sleep-wake phase disorderc. Irregular sleep-wake rhythmd. Non-24-hour sleep-wake rhythm disordere. Shift work disorderf. Jet lag disorderg. Circadian rhythm sleep-wake disorder not otherwise specified (NOS) 5. ParasomniasA parasomnia is an unwanted physical movement or action during sleep. This group of disorders is classified by disorders or arousal from NREM sleep, those associated with REM sleep, and other parasomnias.a. NREM-related parasomniasi. Disorders of arousal from NREM sleepii. Confusional arousalsiii. Sleepwalkingiv. Sleep terrorsv. Sleep-related eating disordersb. REM-related parasomniasi. REM sleep behavior disorderii. Recurrent isolated sleep paralysisiii. Nightmare disorderc. Other parasomniasi. Exploding head syndromeii. Sleep-related hallucinationsiii. Sleep enuresisiv. Parasomnia due to medical disorderv. Parasomnia due to medication or substancevi. Parasomnia, unspecifiedd. Isolated symptoms and normal variantsi. Sleep talking 6. Sleep-related movement disordersThis class of disorders is characterizeed by simple, often repetitive movements during sleep or wake that can disrupt the sleep of the patient, the patient's bed partner, or both.a. Restelss leg syndromeb. Periodic limb movement disorderc.Sleep related leg crampsd. Sleep-related bruxisme. Sleep-related rhythmic movement disorderf. Benign sleep myoclonus of infancyg. Propriospinal myoclonus at sleep onseth. Sleep-related movement disorder due to medical disorderi. Sleep-related movement disorder due to medication or substancej. Sleep-related movement disorder, unspecified
3a) Sleep Disturbances and CFS as sleep disorder
3b) Hypersomnolence, Hypersomnias
3c) Excessive daytime sleepiness (EDS)
3e) Sleep Apnea (obstructive or Central)
3e i) Neurological Effects of Apnea
3e ia) Treatment
3e ib) Cpap, Apap, Spap (etc) machinery
3e ic) Apnea surgery
3e iii) Dental mold sleep positioner device
3e iv) CPAP and surgery alternatives
4) Sleep circuits, Systems and Regions
4a) Reticular Activating System
4b) Ascending Arousal Pathway (ARAS)
4b i) Ascending Arousal Systems in the Regulation of Sleep and Awakefulness Mediated by the Basal Forebrain
4b ii) Neuroanatomic Connectivity of the Ascending Arousal System Critical to Consciousness and Its Disorders
4c) Descending Reticular Activating System (DRAS)
4e) Hypocretin/Orexin System
4f) GABA/galaninergic System
4f i) GABA receptors
4f ii) Modulation of Vigilance in the Primary Hypersomnias by Endogenous Enhancement of GABAA Receptors
4f iii) The relationship between sleep and wakefulness may depend on the balance of activity in the GABA/galaninergic systems and orexin/hypercretin systems of the posterior hypothalamus
4f iv) Histaminergic neurons of the hypothalamic tuberomammillary nucleus constitute a major wake‐promoting system
4g) Somnogen in Cerebrospinal Spinal Fluid of Hypersomnia Patients, and (Endogenous) Enhancement of GABAA Receptors
4h) Hypothalamus
4hi) Myalgic Encephalomyelitis/Chronic Fatigue Syndrome as Disturbed Homeostasis due to Focal Inflammation in the Hypothalamus
4hii) "The sleep switch", hypothalamic control of sleep and wakefulness
4i) Hippocampus
4j) HPA axis
5) Sleep Medicine
5a) Histamino‐mimetic drugs such as H3‐receptor inverse agonists could, in fact, become a treatment option not only in patients with narcolepsy (Lin et al., 2008) but also for those with EDS of other origin
5b) Drugs for narcolepsy
Drugs prescribed for narcolepsy
5c) Treatment with Flumazenil (GABAA receptor antagonist)
5e) Sleep aid drugs
6) CFS similarities analogy to Hibernation state
6a) Cell Danger Response Theory
6b) Best diet for patients affected by CDR
7) Fatigue from TBI and Factors contributing to chronic fatigue after traumatic brain injury
7a) Sleep Disturbance After Traumatic Brain Injury (TBI)
7b) TBI and hormones
7c) Changes in Emotions or Sleep Patterns
7e) Disturbed sleep
Nocturnia
8) Studies done on Sleep Issues (links)
9) Miscellaneous
9a) Sleepiness
9b) Cortisol Mean, Cortisol Slope, or Abnormal Cortisol Levels Were not Related to 9c) Neurocognitive Symptoms, Psychiatric Status, or Sleep Difficulties (just 1 study)
9c) Indirectly sleep related
Indirectly sleep relatedreported deficiencies in the urea and the TCA cycles, (ornithine/citrulline and pyruvate/isocitrate ratios) reported deficiencies in the urea and the TCA cycles, (ornithine/citrulline and pyruvate/isocitrate ratios), which ultimately result in reduced levels of ATP production in patients with ME/CFS (Yamano, et al., 2016). Reduced Substrates That enter oxidation downstream of pyruvate dehydrogenase (PDH) Studies revealed that ME/CFS have reduced substrates that enter oxidation downstream of pyruvate dehydrogenase (PDH), such as glutamine, glutamate and phenylalanine, thus suggesting impaired pyruvate catabolism, which ultimately results in increased utilization of acetyl-CoA-producing amino acids as alternative substrates for fuelling aerobic metabolism via the TCA cycle (Armstrong, et al., 2012), (Armstrong CW, et al., 2015), (Fluge, et al., 2016). Glutamine Phenylalanine Reduced concentrations of amino acids that maintain TCA cycle capacity (suggesting) impaired fuelling of the TCA cycle by pyruvate Reduced concentrations of amino acids that maintain TCA cycle capacity were detected in patients with ME/CFS (Fluge, etal., 2016), suggesting impaired fuelling of the TCA cycle by pyruvate. TCA cycle intermediates reduced in Urea and Plasma Reduced ATP production is associated with increased levels of reactive oxygen species (ROS) The role of mitochondrial dysfunctions due to oxidative and nitrosative stress in the chronic pain or chronic fatigue syndromes and fibromyalgia Increased ROS in CFS and FM, resulting in impaired mitochondrial function and reduced ATP in muscle and neural cells, might lead to chronic widespread pain in these patients. Therefore, targeting increased ROS by antioxidants and targeting the mitochondrial biogenesis could offer a solution for the chronic pain in these patients. The role of exercise therapy in restoring mitochondrial dysfunction remains to be explored, and provides important avenues for future research in this area Reduced ATP production is associated with increased levels of reactive oxygen species (ROS), which may ultimately lead to mitochondrial damage and the hypometabolic profile of ME/CFS (Naviaux, et al., 2016) (Armstrong CW, et al., 2015) Mitochondrial Inhibition, Damage and Hypometabolism ROS (and Ros mediated damage) reduced levels of ATP or ATP production Steroidogenic Pathways Neurosteroidal Systems Steroid Hormones T1T2 cycle DHEA DHEA’s involvement in immune homeostasis is anti-Th2/pro-Th1 steroids cortisol damaging, check oxidatn noroadrenaline the problemcfs modulator of [neurotrophic factor receptors Abnormalities in Cerebral Perfusion Abnormalities of AMPK activation and glucose uptake Effect of EPS on AMPK activation, MHC expression and cytotoxicity
![]()
Fatigue
Oxford Textbook of Neurorehabilitation (2 edition 'The impact of fatigue on neurorehabilitation'
https://oxfordmedicine.com/view/10.1093/med/9780198824954.001.0001/med-9780198824954-chapter-26
The impact of fatigue on neurorehabilitation IntroductionThough difficult to define, and abstract in conceptualization, fatigue is an experience familiar to all. Consequently, it is not itself pathological but is experienced to an extreme and disabling degree in many neurological conditions. In chronic fatigue syndrome (CFS) extreme fatigue is the central feature of the condition, and the physiological changes and behaviours associated with it probably contribute to the pervasiveness of symptoms and chronicity of the problem. These physiological changes and behaviours likely also contribute to fatigue in other conditions. These often represent potentially remediable factors, and consequently are important to identify and address. An individualized formulation is crucial to achieving this, a principle which will guide the structure of this chapter. First, we will consider how factors innate to neurological conditions and those secondary to them can contribute to fatigue. Then, we will discuss how understanding of these factors can be synthesized in the formulation to guide treatment. This chapter will focus on the commonest neurological conditions in which fatigue is prominent; multiple sclerosis (MS), stroke, Parkinson’s disease (PD), and traumatic brain injury (TBI). The same principles are applicable to fatigue in other neurological conditions General issuesWhat is fatigue? The distinction between ‘normal’ and ‘pathological’ fatigue is unavoidably arbitrary. Nonpathological fatigue would be the experience of being tired after exercise, with energy restored after rest [1] . Definitions of pathological fatigue vary, but that of the MS Council’s is a good example, namely: ‘a subjective lack of physical and/or mental energy that is perceived by the individual or caregiver to interfere with usual and desired activities’ [2]. The subjectivity of fatigue is emphasized in this definition, which is consistent with the description of (poststroke) fatigue as ‘weariness unrelated to previous exertion levels and usually not ameliorated by rest’ [3]. Other definitions, such as Staub and Bogousslavsky’s, emphasize the subjective need for greater effort, which does seem to be a core feature of the fatigue experience in all contexts [4 Though attempts have been made to separate physical and mental fatigue, this is often neither feasible nor useful. Others favour dividing fatigue in to ‘central’ and ‘peripheral’ components. Chaudhuri and Behan (2000) for example, define central fatigue as ‘the failure to initiate and/or sustain attentional tasks and physical activities requiring selfmotivation (as opposed to external stimulation)’, whereas peripheral fatigue is regarded as primarily physical or muscular in nature [5] . Central fatigue would be exemplified by the perception that more effort is required to undertake a task than is normal in the absence of any overt physical disability (i.e. a sensory symptom), while peripheral fatigue might be characterized by the objective documentation that voluntary power declines during a task or on repetition of that task (i.e. a motor sign) [6]. Notwithstanding that neurophysiological testing is crucial in distinguishing the level of involvement of the motor system, in practice the usefulness even of this more considered distinction can be questionable [7]. Perceived levels of central and peripheral fatigue covary, and even in myasthenia gravis, that exemplar of ‘peripheral fatigue’, reports of mental fatigue are common [8 Fatigue is dissociable from sleepiness although there is overlapping symptomatology [9] . Sleepiness is a physiological phenomenon, depending on previous sleep and occurring at regular intervals following a circadian rhythm [10]. By contrast, fatigue describes persistent physical or mental exhaustion, not necessarily accompanied by sleepiness, and which sleep cannot alleviate. A patient may have both, and they are associated with common factors (e.g. the release of proinflammatory cytokines) [11]. Apathy, even harder to distinguish from fatigue, is a feeling of indifference; fatigued patients generally retain an interest in their hobbies and interests, they just do not have the energy to undertake them Recently models of the ‘predictive brain’ from computational neuroscience have been applied to try and provide a unifying theory to understand fatigue. These models conceptualize the brain as constantly trying to minimize ‘surprise’ by predicting sensory input while simultaneously driving behaviour to maintain homeostasis. Within this model, Stephan et al. propose fatigue to be a feeling arisen from a state of chronic dyshomeostasis [12]. Essentially, in the context of a chronic perturbation (which in MS for example could be chronically elevated cytokines or dopamine imbalance due to failure of nonmotor functions of the basal ganglia) there is persistent interoceptive surprise which represents a warning to the brain that it cannot maintain homeostasis. This may underpin the experience of fatigue, which as it is accompanied by metacognitive recognition of the failure of homeostatic control strategies, impacts on selfefficacy beliefs and is associated with discomfort. It may drive rest, but as this does not result in resolution of fatigue, predictions are modified with the expectation fatigue will be an enduring state. Consequently, even if the precipitating perturbation does resolve, this expectation can persist. This prediction alters neurocomputational processes during monitoring and preparation of effortful physical activity which results in the ongoing experience of fatigue [13]; the related changes in behaviour continue, further maintaining the fatigued state Complementing the predictive brain model of perception is the idea that the motor cortex generates predictions of the sensory consequences of its output, this ‘efference copy’ alerting sensory cortices to upcoming feedback and changing their response properties. This efferent copy is experienced as a sensory experience, the force developed by a muscular group being perceived indirectly through the effort necessary to generate it [14]. The active inference theory of sensorimotor control expands on this, postulating that for a movement to be initiated (and sensory predictions to be fulfilled), ascending sensory errors must not be attended to, a process known as ‘sensory attenuation’ [15]. In Annapoorna Kuppuswamy’s words, ‘we have to transiently suspend attention to sensory evidence we are not moving’ [16]. If this process of sensory attenuation is impaired, it is argued, these ascending sensory errors are attended to, which is inferred by the brain as more than the estimated effort being required to perform the muscle contraction [16]. It is this which is experienced as fatigue. As expected by the predictive brain model of fatigue, this memory of effortful activities influences resting state spontaneous neuronal firing meaning that over time chronic fatigue is experienced even at rest How to measure a subjective conceptPeripheral fatigue, as an acute tendency for force generating capacity to diminish during sustained effort, can be fairly easily described and quantified. It is attributed to mechanisms such as the failure of neuromuscular transmission, metabolic disturbances, defects of muscle membranes, or peripheral circulatory failure [5] . Central fatigue is subjective, warranting intervention when a patient endorses the symptom, its importance, and desire for treatment. Many attempts have been made to quantify it. Mead et al. identified over 50 scales used to assess fatigue poststroke [17], some of the more commonly encountered being summarized in Table 26.1. Others have taken a different approach to assessing fatigue. For example, in stroke a semistructured interview has been used, and ‘caseness’ defined as fatigue present most days for >50% of waking hours which interferes with everyday activities [18]. This approach has also been used to define CFS. It is clear however that all cutoffs for ‘caseness’ are arbitrary, and as all assessments rely on subjective reporting they do not differentiate inability to generate or maintain the required effort/force from disinclination to do so [6 Attempts have been made to objectively quantify ‘central’ fatigue. In ‘cognitive’ fatigue, likely best conceptualized as one component of it, these have generally measured performance on tasks requiring sustained attention. Meaningful associations between subjective reports of fatigue and neuropsychological measures have been elusive however [19]. This is perhaps unsurprising given the lack of an association between cognitive complaints and performance on neuropsychological assessment [20]. Though an MS study did report an association between subjective fatigue and performance when executive demands were very high [21], and a stroke study related fatigue to attentional and executive impairment [22], objective measures of central fatigue are currently of little clinical utility Another way of categorizing fatigue is dividing it into perceived fatigue and performance fatigability. Perceived fatigue is defined as subjective sensation and measured by questionnaires. Performance fatigability in contrast is a decline of objective measures of physical or cognitive performance over a discreet period of time and measured by physical or cognitive tasks. Use of this approach has been suggested in stroke [23, 24], PD [24], and MS [25]. In most cases, fatigue and fatigability probably coexist. Systematic review of 19 MS studies (n = 848) showed that there is a significant correlation between fatigue and fatigability (r = 0.31; p <0.001). However, the strength of the association between those two constructs was not high enough to conclude that fatigue and fatigability are the same phenomenon and they should be assessed separately [25]. This has implications for practice, as perceived fatigue and fatiguability may respond to different treatment approaches EpidemiologyCommunity studies report a prevalence of debilitating fatigue lasting at least six months of around 5% [26]. Greater fatigue is weakly associated with increasing age, female gender and, possibly, lower socioeconomic status [27, 28]. The prevalence of fatigue in neurological conditions is increased beyond what would be expected solely on the basis of age and disability, being estimated to affect 30–80% of patients (see Table 26.2) [24]. Patients also often report a qualitative difference in their experience of fatigue after acquiring a neurologic illness, describing it as ‘overwhelming’ and ‘mindnumbing’ [29, 30]. Impact of fatigueApproximately 40% of MS and onethird of PD patients report fatigue as their most disabling symptom [31, 32]. Fatigue imposes significant socioeconomic consequences, including loss of work hours and may be the most important factor in loss of employment [33, 34]. Fatigue was identified as one of the top ten research priorities relating to life after stroke, as agreed by stroke survivors, caregivers, and health professionals [35 Aetiology and associationsA combination of biological, psychological, and social factors contributes to fatigue in all patients.Though the relative contribution of each varies between diagnosis and patient, failure to consider each sphere can lead to suboptimal treatment in all. Obvious examples are failure to consider depression in an individual who has prominent fatigue following a stroke, or overlooking medication side effects in a patient with CFS. Psychosocial issues, including unhelpful health beliefs, are particularly important to identify in neurological disease, as these may be the most modifiable maintaining factors. Though divisions are fluid, in this section potential contributions to fatigue will be separated into ‘primary’ and ‘secondary’ factors. The former are directly attributable to the neurological disease process, while the latter are physiological, psychological, or behavioural changes occurring as direct or indirect consequences. CFS is an example of a condition in which extreme fatigue exists in the absence of overt neurological pathology, but the presence of immunological and endocrine abnormalities is well established. This demonstrates both the arbitrary separation between ‘primary’ and ‘secondary’ factors and the potentially profound impact that the latter can have. Effective treatment for CFS suggests what may improve fatigue in other conditions. Primary factorsDirect brain pathologyNeurological disorders are due to abnormalities of the structure or function of the nervous system. Those associated with fatigue affect diverse brain regions however, and fatigue is also prominent in medical and psychiatric conditions in which brain structural abnormalities are subtle or absent. Consequently, fatigue is unlikely to be localized to a discrete brain region Structural MRI studies of MS have reported associations between fatigue and volume loss [36], with atrophy in the striatum, thalamus, frontal, and parietal cortex particularly highlighted [37, 38]. In a 2009 crosssectional study poststroke fatigue was more common in stroke than transient ischaemic attack (TIA) patients, suggesting at least some poststroke fatigue might be attributable to brain damage [39]. Systematic review found no association between fatigue and white matter lesions or brain atrophy however, though some studies did report an association with infratentorial or basal ganglia stroke [40]. No TBI studies examine correlations between structural abnormalities with fatigue, but clinical markers of injury severity do not predict fatigue [41]. A study in patients who had had penetrating TBI found that fatigue was associated with ventromedial prefrontal cortex damage [42]. Reduced grey matter volume is reported in CFS [43], with increased prefrontal cortex volume following treatment with cognitive behavioural therapy (CBT) [44]. In summary, when structural abnormalities are identified they implicate involvement of frontal and subcortical brain regions in fatigue As fatigue likely involves distributed brain regions, functional imaging may provide greater insights into its mechanisms. These approaches generally support the concept that fatigue is associated with damage to cortical–subcortical circuitry, particularly circuits involved in attention and executive function. In MS there is decreased regional glucose metabolism in the frontal cortex and basal ganglia of fatigued patients [45]; in TBI brain activity is increased in the middle frontal lobe, basal ganglia, and anterior cingulate during a speeded cognitive task [46]; in PD decreases in frontal lobe perfusion are greater in patients with fatigue than those without (which was associated with executive function impairments) [47]; and CFS patients had differing patterns of activation of prefrontal cortical regions compared to healthy controls [48]. Functional imaging studies of fatigued stroke patients have not been undertaken, but poststroke fatigue has been related to attentional and executive impairment [22]. Fatigue in PD could be associated with a reduction in serotonergic function in the basal ganglia and limbic structures and insular dopaminergic dysfunction [49 In summary, convergent data across neurological conditions suggest dysfunction in the striatalthalamicfrontal system is important in fatigue. These impairments may necessitate higher levels of mental effort for complex tasks, which increases subjective fatigue. In conditions with damaged brain structure (e.g. MS, TBI) recruitment of expanded pools of cortical neurons likely reflects brain plasticity unmasking latent pathways. Though adaptive, it may be energy intensive, excessive use of neuronal pools resulting in fatigue [50]. Returning to the predictive brain model of fatigue, metacognitive detection of such a general slowing and inefficiency of cognition, may lead to a similar sensation of fatigue as when caused by bodily dyshomeostasis [12]. In CFS disruption again seems present but likely arises through different routes, which may include mechanisms such as sustained abnormalities of attentional focus. Impairments in regulation of bodily states important in maintaining fatigue may be the consequence rather than cause of beliefs about low allostatic selfefficacy. Inflammation and endocrine factorsInflammation is associated with fatigue, as evident from the lethargy of acute infections. This is mediated by proinflammatory cytokines, which act on the brain to result in drowsiness, loss of appetite, decreased activity and withdrawal from social interaction [51]. It is thought that these cytokines drive the enzyme indole2,3dioxygenase (IDO) to transform tryptophan into kynurenine; this means that less tryptophan is available for generation of serotonin (levels of which are reduced), but also results in accumulation of kynurenine [52]. Kynurenine is further catabolized into neuroactive metabolites such as kynurenic acid and quinolinic acid which are neurotoxic and believed to lead to dysfunction of frontostriatal networks and ultimately sickness behaviour [53 The association between treatment with interferon alpha (IFNα) and fatigue (which is dissociable from depression) is of course well recognized [54]. As inflammatory degenerative disorders, elevated cytokines are particularly relevant to fatigue in MS and systemic lupus erythematosus. Serum levels of TNFα, IFNγ and IL6 have all been shown to be elevated in fatigued versus nonfatigued MS patients [55–57]. Cytokines are however also elevated poststroke and TBI [58, 59], in CFS [60], and even in PD [61], and depression [62], likely also contributing to fatigue in these conditions Alterations in the hypothalamicpituitaryadrenal (HPA) axis are among the most replicated findings in CFS, mild hypocortisolaemia being consistently reported and attributed to enhanced negative feedback in the HPA axis [63]. This contrasts with the increased HPA axis activity and raised cortisol levels seen in depression [64]. Whereas hormonal changes are relatively subtle in MS and CFS, in TBI, they can be gross and necessitate replacement treatment; this is the case most obviously in pituitary stroke. In TBI these abnormalities are not restricted to the acute phase, with as many as 25% of longterm survivors showing one or more pituitary hormone deficiencies [65]. As well as hypocortisolaemia and hypothyroidism being obvious causes of fatigue, an association with lowered growth and sex hormone levels following TBI has been reported [66, 67 Pituitary dysfunction is common after TBI and stroke, especially subarachnoid haemorrhage (SAH). According to Booij et al., up to 85% of stroke survivors have some degree of pituitary dysfunction [68]. Growth hormone deficiency was most common and was found in nearly half of patients after TBI or SAH. Data on the association between fatigue and pituitary dysfunction are scarce however and more research is needed Genetic polymorphisms. Most studies examining the relationship between genetic polymorphisms and fatigue have been in CFS. Proposed generelated contributors to fatigue include neurotransmitter dysregulation, changes in activity of the HPA axis, immune dysregulation, and abnormalities in muscle metabolism. Wang et al. recently reviewed associations between single nucleotide polymorphisms (SNPs) and fatigue in CFS, cancer and other medical conditions [69]. SNPs in regulatory pathways of neurotransmitters and the immune system were associated with fatigue in all three groups, with several reports of associations with SNPs in regulatory pathways of the serotonergic system and HPA axis in CFS. One study reported that SNPs could be used to predict CFS with 72–76% accuracy, but to our knowledge this has not been replicated [70 Associations between genetic polymorphisms and fatigue in neurological disease have received little attention. Of six serotoninassociated genes examined, ChoiKwon et al. found a relationship with poststroke fatigue only with a polymorphism associated with low monoamine oxidase A activity—and this association was present only in women [71]. A small, exploratory, poststroke study of two genes involved in inflammatory processes reported that specific genetic polymorphisms associated with higher circulating inflammatory biomarkers (such as IL1β and C reactive protein) were associated with greater selfreported fatigue [72]. Intriguing though these findings are however, the reality is that studies tend to be small and are generally unreplicated; at the present time genetic testing generally has no direct clinical application to fatigue management. Secondary factorsOther medical problemsThe possibility of additional medical pathology must be remembered. There should be blood screens for common haematologic and metabolic conditions and thyroid dysfunction. Recommended investigations to aid diagnosis of CFS, exclude other conditions and ‘red flags’ for alternative diagnostic explanations are shown in Box 26.1 [73]. In MS vitamin D deficiency is common. An association with fatigue has been found in the general population and a randomized controlled trial (RCT) has reported fatigue improved on treatment with cholecalciferol [74]. Though an RCT found cholecalciferol did not reduce fatigue in patients with relapsing remitting MS being treated with IFN, patients were not selected on the basis of vitamin D deficiency [75]. In fatigued patients it seems reasonable to consider checking 25hydroxy vitamin D levels, initiating treating if low. Infections can worsen fatigue and should be excluded [76]. An MS exacerbation may present as fatigue prior to clinical manifestation [77]. Medication side effects Medications frequently causing fatigue include antispasticity agents (e.g. Baclofen or Tizanidine), narcotic analgesics, sedative hypnotic, or anticonvulsant agents, sedative antidepressants or anxiolytics and antihypertensive medication [78]. Patients often report increased fatigue with interferon therapy, though fatigue often improves with time on interferon [79]. Pretreating with nonsteroidal antiinflammatory may improve IFN associated fatigue [78]. Hypertension or hypotension secondary to excessive antihypertensive use may be associated with poststroke fatigue, though whether there is a causal relationship is uncertain [80]. It is unlikely that poststroke fatigue could be a mere side effect of medications, but polypharmacy can certainly increase the likelihood of it developing [81 Mobility issues and environment In stroke, TBI and MS ambulation can be compromised by spasticity and weakness. Gait can be inefficient, requiring excessive energy expenditure that quickly fatigues the patient [82]. This will reduce physical activity causing physical deconditioning. The oxygen cost of breathing is increased in PD patients, meaning they require more energy simply to breath [83]. Energy loss with tremors and dyskinesias has also been demonstrated [84]. Hyperthermia contributes to fatigue during exercise in healthy people [85], but the strong association between heat and symptom exacerbation (Uhthoff’s phenomenon) is a particular characteristic of MS. It affects 60–80% of MS patients and is attributed to increased body temperature inducing conduction block in vulnerable axons [86]. Deconditioning can result through avoidance of exercise/activity to prevent symptom exacerbation [87]. Psychiatric conditionsFatigue and depression after stroke have a bidirectional association (i.e. patients with fatigue are more depressed and patients with depression are more fatigued) [88]. Fatigue is a core feature of depression, present in around 25% of people with neurological disorder [89]. Depression severity correlates with fatigue severity [90], and should never be dismissed as simply ‘an appropriate reaction to a serious illness’. Indeed, rather than being ‘secondary’ to neurological conditions its aetiology is often not clearly separable from the neurological disease process itself. A positive correlation has also been reported between anxiety and subjective fatigue [91]. Identification of depression in neurological conditions is complicated by many symptoms (e.g. fatigue, reduced attention and concentration, disturbed sleep) being features of the diseases themselves. Consequently, greater emphasis should be placed on cognitive than somatic symptoms, the presence of guilt, worthlessness, hopelessness, and suicidality strongly suggesting depression. The pattern of fatigue observed in depression is rather different from that in neurological conditions. Fatigue in depression tends to be worst in the morning, improve as the day goes on, and not be relieved by sleep; MS fatigue by contrast is best in the morning, worsens as the day goes on, and rest gives some relief. Substance misuse is strongly associated with fatigue and should always be considered. Anxiety is less studied than depression, but in the Nottingham Fatigue After Stroke study (which excluded participants with high levels of depressive symptoms) was found to be strongly associated with fatigue at both 4–6 weeks and 6 months poststroke [92, 93]. It is clearly conceivable that anxiety, with its associated increase in threatrelated attention, could both intensify the perception of fatigue and drive avoidance and deconditioning. Sleep disordersIn neurological disorders fatigue has a consistent relationship with broken sleep. Correlations with daytime sleepiness however, though present in TBI and PD, are surprisingly weak in MS [94–96]. Nonetheless, patients reporting daytime sleepiness should be screened for potential sleep disorders, including obstructive sleep apnoea, narcolepsy, and restless leg syndrome/periodic limb movement disorder. The latter are actually very common in MS [97], while sleep disordered breathing is a particular issue after stroke [98], and various sleep disorders are core features of PD [99]. Initial insomnia in the absence of obvious medical cause suggests anxiety, while early morning waking with inability to get back to sleep is more characteristic of depression. Other diseaseassociated problems such as pain, spasticity, and nocturnal micturition also impact on sleep and require specific interventions. Pain Pain is common and can be difficult to treat. Robust correlations between pain and fatigue are reported in various neurological conditions [100, 101]. It is speculated that pain may contribute to fatigue through a reduction in central motor drive [102]. If associated with activity it will encourage activity avoidance. About 10% of patients after stroke present with triad of pain, fatigue, and depression and 20% pain and fatigue [101]. Poor nutritionAs 50–80% of inhospital stroke patients have one or more eating difficulties related to neurologic deficits, it is unsurprising that 50% of stroke inpatients are malnourished [103]. Westergren reported that six months poststroke poorer nutritional status was closely related to a lack of energy [104]. Effects are likely bidirectional, giving rise to a vicious circle. DeconditioningDeconditioning is a complex physiological process in which the lack of use of the body’s cardiovascular, neuromuscular, biomechanical, and musculoskeletal systems leads to a decrease in their functional capacity, and the body’s efficiency [105]. This reduces the capacity for exercise and increase the perception of effort required for a given level of activity. Though evidence of significantly reduced physical fitness in CFS compared to sedentary controls is conflicting, a systematic review concluded that there was reduced physiological exercise capacity in CFS and that deconditioning is a perpetuating factor [106]. Though other factors likely contribute too, some evidence for the presence of deconditioning is provided by the proven efficacy of graded exercise programmes in reducing symptoms of CFS [107]. Neurological conditions are associated with substantial reductions in activity, and deconditioning has been reported in MS, stroke, TBI, and in PD [108–111]. Beliefs about activity/cognitive style Cognitive factors believed to contribute to the maintenance of CFS include a tendency to focus on fatigue and perceive it as a negative experience (which consequently amplifies its perception), beliefs of having a very limited ability to be active (leading to very low levels of activity) and expectations of chronicity (which fits with the ‘predictive brain’ model of expectation determining perception) [112, 113]. In CFS a strong belief in a physical cause of the illness, a strong focus on bodily sensations, and a poor sense of control over symptoms contribute to fatigue severity and functional impairment [114]. The importance of fear of symptom exacerbation in CFS was elegantly demonstrated in a cycling task. It was a more important determinant of distance travelled than physical symptoms, physical disability, mood, and other illness perceptions [115]. It is likely that similar cognitions and attentional biases magnify fatigue in some with neurological disorders. Beliefs about the dangers of activity may be expected in MS, given that elevated body temperature or prolonged exertion exacerbates symptoms [78]. Focusing on personal emotions, selfblame, and maladaptive coping (e.g. denial and selfdistraction) have been associated with poststroke fatigue [116]. Though unstudied, it is likely that after a catastrophic event such as stroke fears of provoking a further stroke inhibit engagement with exercise in some patients. It has been reported in MS that a sense of control, (often referred to as selfefficacy), reduces feelings of fatigue, whereas focusing on bodily sensations aggravates it [117]. MS patients who catastrophize about experiencing symptoms (expecting the worst possible outcome), who are embarrassed about symptoms, or who believe symptoms are always a sign of physical damage are more likely to be fatigued [118 A systematic review incorporating studies in cancer, CFS, MS, fibromyalgia and healthy individuals confirmed a significant association between catastrophizing and fatigue [119]. Other cognitive styles also influence fatigue and activity levels. For example, a qualitative study examining what determined whether people resumed previously valued activities after stroke identified ‘allornothing’ thinking as a barrier [120]. ‘Allornothing behaviour’ (i.e. overdoing things when feeling better then needing to rest for prolonged periods to recover) is associated with fatigue in MS [118]. Preinjury factors and response to stressorsPersonality is assumed to influence vulnerability to CFS, but crosssectional studies confound state and trait effects. A prospective study did however report that higher emotional instability (an individual’s tendency to experience psychological distress) and selfreported stress were risk factors for the condition [121]. Acute physical or psychological stress might trigger the onset of CFS, it being shown that severe stressful events or difficulties are more common in the period prior to onset of the illness than in population controls [122]. Having a severe neurological disorder is a significant stressor, and dispositional differences in response to stress must contribute to fatigue in at least some patients. Though prospective studies are lacking, crosssectional data reports higher emotional instability is associated with greater fatigue in MS and cancer [123, 124] and neuroticism and fatigue after stroke [125]. An association between fatigue and perceived stress has also been reported [126]. In a cohort which excluded participants with substantial depressive symptoms, prestroke fatigue increased the risk of poststroke fatigue; indeed (together with having a spouse or partner, lower Rivermead Mobility Index score, and higher depressive and anxiety symptomatology), prestroke fatigue accounted for approximately 47% of the variance in Fatigue Severity Scale scores at 4–6 weeks poststroke [92]. Assessment of prestroke fatigue is obviously retrospective and susceptible to recall bias, however [127], and the picture further complicated by fatigue itself being a risk factor for stroke [127 TreatmentThe multifactorial nature of fatigue is captured by a biopsychosocial formulation, which facilitates an individualized understanding of maintaining factors and guides multidisciplinary management. A model formulation is depicted in Fig. 26.1. Unfortunately, the evidence base for the treatment of fatigue in neurological conditions is poor. Most research on fatigue treatment has been in CFS and MS, so evidence will often be extrapolated from these conditions. The absence of overt neurological pathology in CFS means this must be done with caution. However, as discussed earlier, there is considerable overlap between biological, psychological, and behavioural findings in fatigued individuals across diagnoses, which provides some justification. ‘Secondary’ factors certainly contribute substantially to fatigue in neurological conditions, and being generally more modifiable than the disease process itself are important treatment targets. Review treatment of neurological condition and directly related problemsThough fatigue is a side effect of some immunomodulatory agents used to treat MS, diseasemodifying drugs can actually reduce fatigue. This is reported even with IFNβ, but the effect with Glatiramer Acetate is significantly greater [128]. A crosssectional casecontrolled study suggested Natalizumab may have greatest effect [129]. There is not strong evidence that teriflunomide or dimethylfumarate improve MS fatigue, though patient ratings with both treatments compare favourably to IFNβ [130, 131]. PD drugs are implicated both in exacerbating and reducing fatigue. Pramipexole has been associated with increased subjective fatigue in several RCTs [132], while Carbidopa–levodopa reduced muscle fatigue in experimental studies [133]. Two RCTs reported a beneficial impact of the MAOB inhibitor rasagiline on fatigue in patients with PD [134, 135]. That of Stocchi et al. was by far the largest (n = 1176) and reported less progression of fatigue in recently diagnosed PD patients treated with rasagiline 1 mg or 2 mg when compared to placebo over 9 months [135]. The study evaluated the agent as a diseasemodifying drug and the authors caution that reducing progression of fatigue does not necessarily equate to ‘treatment’. Assessing subjective fatigue is further complicated in PD by whether the patient is in an ‘on’ or ‘off’ sate. Stroke and TBI do not currently have ‘direct’ treatments, but some symptomatic or secondary prevention interventions are associated with fatigue Identify and treat medical comorbidity and full medication reviewComorbid medical conditions are common in neurological disorders, and are almost universally associated with fatigue Sleep and nutritionSleepSpecific sleep disorders should be identified and treated. Sleep phase disorders are addressed by entraining a regular sleepwake cycle, and melatonin and/or light treatment may assist. CBT has good evidence in treating insomnia in the nonneurological population, with positive studies in TBI and MS [136, 137]. Though hypnotics and sedative antidepressants can be helpful, the high incidence of side effects (falls, hallucinations, sedation, cognitive deficits, bowel and bladder problems, etc.) necessitates caution. Personal experience suggests trazodone and mirtazapine as sedative antidepressants associated with fewest problems, and if they must be used short acting hypnotics (used short term) are preferable to long acting ones. First line treatment of periodic limb movement disorder (PLMD) is reassurance, though dopamine agonists or levodopa can help if treatment is required [138]. Marked relief with ropinirole or pramipexole is reported in poststroke restless leg syndrome (RLS) [139], though ferritin levels should always be checked (and iron deficiency corrected) before starting treatment. Though significant depression must be treated, remember that antidepressants can aggravate RLS and PLMD [98]. Deep brain stimulation has been recommended for the treatment of insomnia in advanced PD [140]. NutritionAll patients should be supported in having a balanced, healthy diet. Even skipping breakfast has been associated with fatigue [141]. In MS, RCTs report adherence to a low fat, low cholesterol diet supplemented with olive oil capsules significantly reduce fatigue [142], but vitamin D or omega3fatty acids do not [143, 144]. Carnitine contributes to cellular energy metabolism, and deficiency may reduce energy production through impaired fatty acid oxidation. An RCT of fatigued elderly individuals without current significant medical morbidity found 4 g a day of acetyl Lcarnitine for 180 days reduced physical and mental fatigue compared to placebo [145]. In an open label randomized study in CFS acetylcarnitine and propionylcarnitine both reduced fatigue, though improvement was reduced with combined treatment [146]. A small crossover randomized trial found acetyl Lcarnitine 2 g daily was associated with greater reduction in MS fatigue (measured by the Fatigue Severity Scale (FSS)) than amantadine 200 mg [147]; in a subsequent study however only amantadine performed better than placebo [148]. Despite reported benefit in various medical conditions, the impact of carnitine on fatigue has not been examined in other neurological conditions. Physical aids and interventionsPhysical aids Use of orthoses or functional electrical stimulators can improve gait mechanics, promote energy conservation, and improve the safety of walking [78], but evidence they reduce fatigue is lacking. Temperature controlGiven the association between heat stress and MS symptom deterioration, simple strategies to minimize heat exposure, such as performing work or exercise during the early morning or late evening when it is cooler, seem sensible. Observational studies report benefit from simple cooling strategies such as cold showers, applying ice packs, and drinking cold beverages [87]. Precooling, essentially immersing the lower limbs in cool water, was shown to have some benefit in terms of walk performance and fatigue ratings [149]. In an RCT, cooling garments demonstrated subjective reductions in fatigue in thermally sensitive MS patients, though blinding of patients was not achieved [140]. Anecdotal evidence suggests 4Aminopyridine limits worsening of MS symptoms during heat exposure or exercise [87]. Treatment of psychiatric conditionsIdentification and treatment of depression and anxiety is crucial, but beyond the scope of this chapter. If insomnia is a major problem sedative antidepressants may be preferable, whereas energizing antidepressants, (such as selective serotonin reuptake inhibitors), may be first choice if fatigue is prominent but initial insomnia not a major issue [150]. Unfortunately, and likely reflecting the multifactorial nature of fatigue, cancer research shows that antidepressant treatment may not improve fatigue even when it treats depression [151]. There is no evidence antidepressants improve fatigue in the absence of depression [152]. Exercise and energy conservation strategiesExercise Exercise improves exercise tolerance and reduces fatigue in healthy individuals as well as those with longterm conditions [153]. Benefit is however not limited to the physical dimension of fatigue, as exercise also improves mood, reduces anxiety and fear [154], and improves cognitive performance [155]. These benefits are likely consequent to associated physiological and anatomical changes, such as increased production of growth factors, increased efficiency of the cerebral vascular system, enhanced hippocampal neurogenesis, and regulation of the immune and endocrine systems [1] . Understanding of how this occurs is fast increasing, a recent study showing voluntary wheelrunning induced gene expression in the mouse hippocampus [155]. As well as addressing deconditioning through these overtly neurobiological mechanisms, exercise gradually allows patients to experience mastery and restore selfefficacy and likely influences effort perception Andreasen et al. categorized exercise interventions as endurance training (ET), resistance training, combined training (CT), and ‘other’ training modalities (OT) [156]. In general ET, or aerobic exercise, is most consistently beneficial. Walking is especially recommended, but swimming or cycling may also be appropriate [157]. Exercise programmes must be properly planned, gradually building levels of activity to promote increasing stamina and prevent unhelpful ‘boombust cycles’ occurring. This means regular exercise sessions start from a level that does not result in postexertional malaise, with the length and frequency of the exercise sessions progressively increasing. The CFS literature shows that, compared with a symptomcontingent approach, a timecontingent approach leads to greater improvements in fatigue and physical functioning [157]. A measurement (time, distance walked, etc.) rather than symptom experience should determine whether a session ends, this likely underpinning the superiority of graded exercise over adaptive pacing [107]. Though patients may be anxious about undertaking an exercise programme, there is no reason why graded exercise carried out under appropriate professional supervision should be harmful. Research on the efficacy of exercisebased interventions in specific conditions will be discussed next Chronic fatigue syndromeA 2011 metaanalysis of five RCTs supported the efficacy of graded exercise in the treatment of CFS [158]. Since this the results of the PACE trial, the largest (n = 640) and most important trial of graded exercise therapy (GET), CBT, and adaptive pacing in CFS have been published. It reported that CBT and GET both had an odds ratio for trial recovery compared to standard care or adaptive pacing of around 3.5, and concluded they ‘can safely be added to specialist medical care to moderately improve outcomes for CFS’ [107]. Concerns about the safety of GET are not evidence based [159]. The underlying principle of gradually increasing activity may be applicable to cognitive activity too, implying CFS sufferers should be encouraged to undertake gradually more challenging intellectual tasks, starting from a tolerable level [157 Multiple sclerosisA recent systematic review evaluated the impact of various psychological interventions on fatigue; these including CBT, relaxation, and mindfulness. CBT was found to significantly reduce fatigue, this effect being evident compared to both treatment as usual (five studies, SMD: −0.32; 95% CI: −0.63 to −0.01) and active controls (three studies, pooled SMD −0.71, 95% CI: −1.05 to −0.37) [173]. Interestingly, though a pilot study, of the studies comparing CBT to treatment as usual the largest effect was actually seen with a telephonesupported internetbased version of this intervention, suggesting this may be a viable costeffective option [174]. The CBT model utilized in the three studies with an active comparator could be rather eclectic, but the largest combined explanation of the cognitive behavioural model of fatigue, activity scheduling, sleep hygiene, and changing unhelpful cognitions about fatigue [175]. It reported that fatigue decreased with both CBT and the relaxation control, but reductions were greater with CBT (effect size 3.0 vs. 1.8). Changing perceptions of fatigue (e.g. perceiving it as more controllable, as time limited and as having less serious consequences) mediated the decreases in fatigue [176]. The same metaanalysis reported that compared to nonactive controls, relaxation training (specifically progressive muscle relaxation) reduced fatigue (two studies, n = 110, SMD = −0.90; 95% CI: −1.30 to −0.51); it is worth noting however that in one of these small studies it was actually outperformed by reflexology [177]. In a relatively large study (n = 150) mindfulness training reduced fatigue [178], a finding replicated in two subsequent RCTs [179, 180]. Mindfulness aims to reduce stress through teaching a nonjudgemental awareness of momenttomoment experience. StrokeAs discussed Zedlitz et al. reported that, though CBT alone had beneficial effects, reductions in poststroke fatigue were greater when cognitive therapy was augmented with graded activity training [164]. The cognitive component was delivered in small groups and emphasized pacing, improved planning of activities and relaxation, with rather less focus on challenging unhelpful cognitions than CBT in the CFS and MS trials. A subsequent small RCT of more ‘typical’ CBT (n = 15), but including input from an exercise physiologist, also reported a positive effect [181]. These results need replication, but do emphasize the importance of exercise/graded activity accompanying any cognitive intervention for poststroke fatigue. A small uncontrolled pilot study suggested mindfulnessinformed CBT was associated with a reduction in poststroke fatigue [182], with a subsequent small RCT (n = 29) of mixed stroke and (severity unspecified) TBI patients also suggesting benefit for mental fatigue [183]. Parkinson’s diseaseA small RCT (n = 28) of computerized CBT found that thought it did benefit the target symptom, insomnia, there was no reduction in fatigue as measured by the FSS [184]. Multidisciplinary rehabilitationMultidisciplinary rehabilitation aims to reduce symptoms, increase independence, and maximize participation in society [185]. It is generally coordinated by a specialist doctor, delivered by a team of different therapists, and not protocolized, being tailored to individual needs and goals. Several uncontrolled studies suggested multidisciplinary rehabilitation is beneficial in CFS [186]. Though impact on fatigue was not a specific focus in Khan et al.’s Cochrane review of the efficacy of multidisciplinary rehabilitation in MS, the review does summarize outcomes on this measure [185]. On the basis of two positive RCTs of outpatient rehabilitation they report there is limited evidence that high intensity programmes can provide shortterm benefit in fatigue, and insufficient evidence that a lower intensity programme can reduce fatigue. A homebased intervention had no impact on fatigue compared to standard care, and the only study comparing inpatient rehabilitation to standard care was negative. This review is being updated [187]. We identified one subsequent relevant RCT which found that outpatient multidisciplinary team input (consisting of physiotherapy, occupational therapy and/or social work input) was not associated with improvement in fatigue relative to the control condition of MSnurse consultation in chronically fatigued MS patients [188]. As the authors acknowledge however advice from MS nurses overlapped with that from therapists, was often quite intensive (mean input 163 minutes vs. 280 minutes) and more medication changes were made in the control group (in 39% vs. 19% of patients). A mediumsized RCT (n = 122) in CFS reported that outpatient multidisciplinary rehabilitation outperformed CBT on assessments performed both after treatment completion and at 26week followup [189]. Though benefits of multidisciplinary rehabilitation have been reported in other neurological conditions, specific effects on fatigue have not been examined. The potentially modest impact of multidisciplinary rehabilitation on fatigue in MS is initially surprising, as it likely includes elements of exercise treatment and cognitive interventions known to have good effect. This may reflect however that it is the delivery of evidencebased interventions directed at specific areas of need that is important rather than simply access to a variety of professionals [190]. MedicationDrugs such as antidepressants have proven benefit in treating secondary conditions common in neurological disease which contribute to fatigue. This section will focus on drugs used specifically to treat fatigue. AmantadineA recent metaanalysis reported that amantadine did reduce fatigue as measured by the FSS (SMD −1.09, CI −1.30 to −0.87), though levels of heterogeneity between included studies was high (I2 = 91%) [191]. It is the only pharmacological treatment for MSrelated fatigue recommended by the UK National Institute for Clinical Excellence [161]. The standard dose is 100–200 mg morning and early afternoon [150]. It is generally well tolerated but side effects can include hallucinations, vivid dreams, nausea, hyperactivity, anxiety, insomnia, constipation, and rash. We could identify no published trials examining the effects of amantadine on fatigue in stroke and TBI, but it was reportedly well tolerated (and effective) when trialled for irritability in the latter [192]. It did not reduce fatigue in a PD study which assessed this as a secondary outcome [193]; the risk of inducing psychotic symptoms is markedly elevated in PD. ModafinilModafinil is a nonamphetaminelike drug approved for the management of narcolepsy which has been used for daytime fatigue in other conditions. A metaanalysis of data from five RCTs (n = 303) showed that modafinil was superior to placebo in patients with MS according to Multidimensional Fatigue Inventory (MFI), while for the FSS there was no significant difference [194]. A small RCT reported improved poststroke fatigue as measured by the FSS (but not the MFI, which was the primary endpoint) [195]. A subsequent large RCT (n = 232) reported significant reductions in poststroke fatigue as measured by both scales after 6 weeks’ treatment with Modafinil 200 mg [196]. Two small RCTs examined the impact of Modafinil on PD fatigue. Though both did not report significant change on any of the FSSs used, one did report improvement in clinical global impression of fatigue [197]; no safety concerns were raised. A metaanalysis of two RCTs (n = 115), showed a therapeutic effect of modafinil on fatigue in TBI (measured by the FSS with a mean difference of –0.82 (95%CI –1.54 to –0.11 p = 0.02, I2= 0%) [198]. Modafinil is generally well tolerated, but serious, lifethreatening skin reactions, psychiatric adverse reactions (such as suicidal thoughts, depression, psychotic episodes) and cardiovascular adverse reactions (e.g. hypertension) have been reported [150]; these, together with the conflicting evidence of benefit, led the European Medicines Agency to conclude in 2011 that the benefits outweighed the risks only for narcolepsy. NICE does not recommend it for MS fatigue [192 Other pharmacological agentsTwo RCTs in MS did not separate effects of Pemoline from placebo [150]; this, together with potential for liver toxicity, mean it is rarely used. Prokarin is a proprietary blend of histamine and caffeine, administered as a cream. A single small MS RCT suggested it reduced fatigue compared to placebo [199]. A single small RCT reported that Methylphenidate 10 mg three times a day significantly reduced fatigue in PD and was well tolerated [200]. It has not been evaluated in other neurological conditions. Lack of replication of this study together with the fact it is a controlled drug, may worsen motor function and quality of life in PD, has a potential for abuse, and can cause insomnia, hypertension, and anorexia, have limited its use. An unreplicated randomized placebocontrolled crossover trial showed benefit from aspirin, daily dosage of 1300 mg, on MSrelated fatigue [201]. 4aminopyridine (Fampridine) does not seem to have a significant impact on MSrelated fatigue [202], despite the fact that good quality studies do report a beneficial impact on walking speed [203]. In a small pilot study (n =12) 10 mg at night of Doxepin, a tricyclic antidepressant, decreased fatigue as measured by the FSSS [204]; this study did not correct for multiple comparisons however, and any benefit could have been driven by improvements in insomnia. Other interventionsSeveral studies have examined the impact of transcranial direct current stimulation (tDCS) on MS fatigue. The most robust of these, targeting the left dorsolateral prefrontal cortex (DLPFC), reported benefit for performancebased measures of cognitive fatigue (it eliminated fatiguerelated increases in reaction time), but no effect on subjective fatigue ratings [205]. Claims have been made for acupuncture in MS fatigue, but the poor quality of studies make this difficult to recommend [206]. Morning treatment with 45 minutes of blue light therapy was found to reduce fatigue in a small RCT of TBI patients (n = 30); improvement was also seen in daytime sleepiness, which may have driven the effect [207]. Benefit was not seen with yellow light therapy and effects were lost on cessation of treatment. Though bright light helped daytime sleepiness in PD, improvements in fatigue were not seen [208]. A pragmatic approach to treating fatigue in neurological conditions This is detailed in Fig. 26.2. Exercise has the best evidence and should be encouraged in all patients. Ideally individually tailored programmes would be provided (e.g. the Exercise After Stroke programme delivered in some UK leisure centres). If not available, but mobility is reasonable, regular walks starting from a modest base are a good option. If there seem to be barriers/reservations about exercise, clinicians must explore what these are and correct any misunderstandings about the perceived dangers of activity/ exercise. CBT has reasonable evidence, but availability is limited. Additionally, many neurological conditions are associated with significant cognitive impairment, which likely reduces the benefit of CBT. Though evidence for pharmacological treatments is weak, amantadine and modafinil have been used. Given the position of the European Medicines Agency and NICE recommendations for the treatment of MS fatigue, the former should be regarded as first choice, though with particular cautions in PD. Carnitine may hold some promise, but further trials are wanted 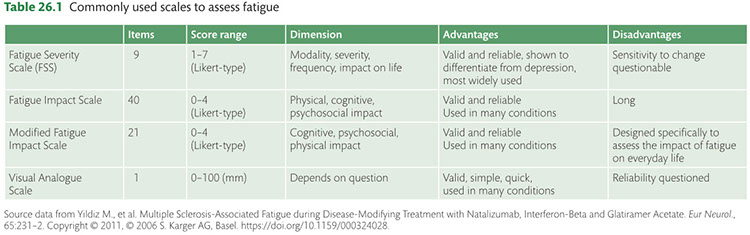
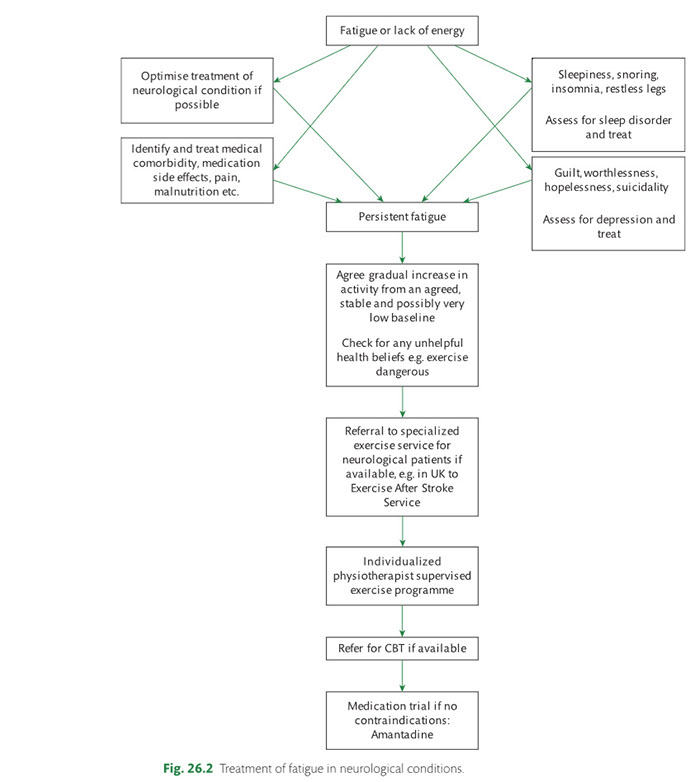
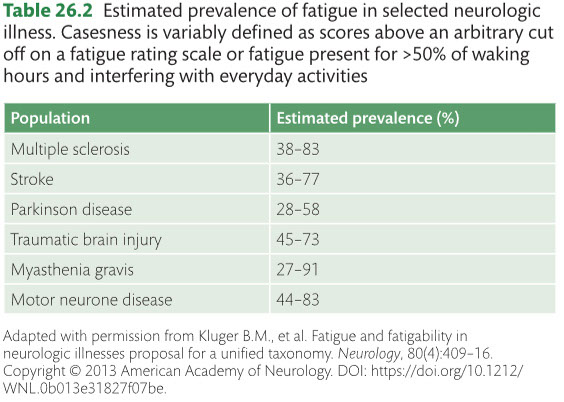

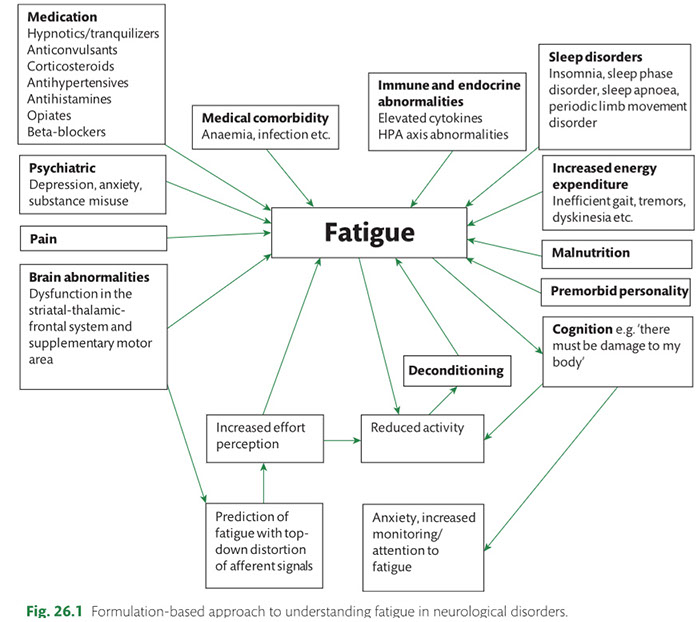 click here to closeback to top of article
click here to closeback to top of article
Recomended 7 to 8 hours a night, every hour of sleep before midnight is worth 3 hours after 10
![]()
I have long considered CFS a sleep related disorder. Most of my research was focusing on finding the hormonal signal(s) to the brain that tells the body to sleep. It is fascinating to discover, as per the research by Naviaux, that the signal resides not in the brain but in the blood itself!
Here I begin this page by examining Hibernation, Torpor, Dauer State and Cell Danger Response as it triggers a switch from aerobic to anaorobic metabolism in chronic fatigue syndrome patients, what induces the signals to the brain for this metabolic switch, and what can be done to switch it back
Eliminate the 'dangers' in the environment that trigger Cell Danger Response (CDR), such as:
Environmental stressors and toxins
Psychological and Emotional stressors and triggers
Eliminate the 'dangers' in the internal environment that trigger Cell Danger Response (CDR), such as:
Latent HHV6
Find a method to induce or encourage a switch back to aerobic metabolism such as:
antipuerinetic medicine
TCM
While waiting until such methods are found, to implement the best methods to subsist on anarobic metabolism such as:
High fat diet
Stress reduction
3a) Somnogen in Cerebrospinal Spinal Fluid of Hypersomnia Patients (click to open)
Somnogen in Cerebrospinal Spinal Fluid of Hypersomnia Patients, and Endogenous Enhancement of GABAA Receptors
Emory Researchers Announce Discovery of Substance That May Be Linked to Excessive Sleeping
https://www.hypersomniafoundation.org/emory-researchers-announce-discovery-of-substance-that-may-be-linked-to-excessive-sleepingDr. David Rye and a team of researchers from Emory University announced their discovery of a substance in the cerebrospinal fluid https://www.hypersomniafoundation.org/glossary/cerebrospinal-fluid of 32 people with idiopathic hypersomnia https://www.hypersomniafoundation.org/glossary/idiopathic-hypersomnia that may be responsible for their relentless need to sleep. For now, the researchers are simply referring to this substance as a somnogen https://www.hypersomniafoundation.org/glossary/somnogen or sleep-producing agent, which appears to heighten the effect of GABA https://www.hypersomniafoundation.org/glossary/gaba when tested in the laboratory. Seven of the research subjects with idiopathic hypersomnia subsequently received flumazenil under experimental conditions and experienced normal levels of alertness.
Modulation of Vigilance in the Primary Hypersomnias by Endogenous Enhancement of GABAA Receptors | Science Translational Medicine http://stm.sciencemag.org/content/4/161/161ra151
The biology underlying excessive daytime sleepiness hypersomnolence is incompletely understood. After excluding known causes of sleepiness in 32 hypersomnolent patients, we showed that, in the presence of 10 μM γ-aminobutyric acid GABA, cerebrospinal fluid CSF from these subjects stimulated GABAA receptor function in vitro by 84.0 ± 40.7% SD relative to the 35.8 ± 7.5% SD stimulation obtained with CSF from control subjects Student’s *ttest, *t*= 6.47, *P< 0.0001; CSF alone had no effect on GABAA signaling. The bioactive CSF component had a mass of 500 to 3000 daltons and was neutralized by trypsin. Enhancement was greater for α2 subunit– versus α1 subunit–containing GABAA receptors and negligible for α4 subunit–containing ones. CSF samples from hypersomnolent patients also modestly enhanced benzodiazepine BZD–insensitive GABAA receptors and did not competitively displace BZDs from human brain tissue. Flumazenil—a drug that is generally believed to antagonize the sedative-hypnotic actions of BZDs only at the classical BZD-binding domain in GABAA receptors and to lack intrinsic activity—nevertheless reversed enhancement of GABAAsignaling by hypersomnolent CSF in vitro. Furthermore, flumazenil normalized vigilance in seven hypersomnolent patients. We conclude that a naturally occurring substance in CSF augments inhibitory GABA signaling, thus revealing a new pathophysiology associated with excessive daytime sleepiness.
http://www.ncbi.nlm.nih.gov/pubmed/?term=Modulation+of+Vigilance+in+the+Primary+Hypersomnias+by+Endogenous+Enhancement+of+GABAA+Receptors published in the November 21, 2012, edition of Science Translational Medicine
Rye DB, Bliwise DL, Parker K, et al. Modulation of vigilancehttps://www.hypersomniafoundation.org/glossary/vigilance/ in the primary hypersomniashttps://www.hypersomniafoundation.org/glossary/hypersomnia/ by endogenoushttps://www.hypersomniafoundation.org/glossary/endogenous/enhancement of GABAA receptors. Sci Transl Med. 2012 Nov 21;4161:161ra151. The paperhttp://www.ncbi.nlm.nih.gov/pubmed/?term=Modulation+of+Vigilance+in+the+Primary+Hypersomnias+by+Endogenous+Enhancement+of+GABAA+Receptors is available free of charge after registering on the AAAS web sitehttp://stm.sciencemag.org/.
Greg Miller, science writer for AAAS, summarized the findings of the paper in ScienceNow in “Putting Themselves to Sleep.http://news.sciencemag.org/2012/11/putting-themselves-sleep”
An Editor’s Summary of Dr. Rye’s paper, “Awake and Refreshed,” was published simultaneously in Science Translational Medicine.
The announcement of this groundbreaking work resulted in a great deal of media coverage in November 2012, some of which is linked below.
Emory University –!Waking Sleeping Beauty: An Antidote for Hypersomniahttps://img.youtube.com/vi/V9gnvWtta4M&feature=relmfu/maxresdefault.jpghttps://youtu.be/V9gnvWtta4M&feature=relmfu
Wall Street Journal – Unraveling the riddle of too much sleephttp://online.wsj.com/article/SB10001424127887324478304578171221462816296.html
National Geographic – Phenomena: Re-Awakeningshttp://phenomena.nationalgeographic.com/2012/11/22/re-awakenings/
MSNBC/Today Show – Disorder causes lawyer to sleep up to 18 hours a dayhttp://video.today.msnbc.msn.com/today/49963811#49963811
CNN/Jane Velez-Mitchell – Anna Sumner Sleeps Up To 18 Hours a Dayhttp://writingshares.com/cnn-jane-velez-mitchell-medical-news-video-anna-sumner-sleeps-up-to-18-hours-a-day-due-to-hypersomnia/
ABC News – Medical Mystery: Body’s Own ‘Valium’ Leads to Extreme Sleepinesshttp://abcnews.go.com/Health/Sleep/medical-mystery-bodys-valium-leads-extreme-sleepiness/story?id=17778685#.UK1Ww4ZuItV
US News & World Report Spinal Fluid Substance May Help Drive Sleep Disorder: Studyhttp://health.usnews.com/health-news/news/articles/2012/11/21/spinal-fluid-substance-may-help-drive-sleep-disorder-study
NBC Atlanta Sleeping Beauty disease affects 400,000http://www.11alive.com/news/article/264935/40/Discovery-of-new-disease-for-people-who-sleep-too-much
Los Angeles Times For the continuously sleepy, a new treatment shows promisehttp://www.latimes.com/news/science/sciencenow/la-sci-sn-continuously-sleepy-treatment-shows-promise-20121121,0,5849003.story
Business Insider – Mystery Substance in the Brain Could Explain Why You’re Always Sleepyhttp://www.businessinsider.com/mysterious-substance-in-brain-of-hypersomnia-patients-2012-11
Deutschlandfunk Aufwachen, bittehttp://www.deutschlandfunk.de/aufwachen-bitte.676.de.html?dram:article_id=230005
The relationship between sleep and wakefulness may depend on the balance of activity in the GABA/galaninergic systems and orexin/hypercretin systems of the posterior hypothalamus
3b) The relationship between sleep and wakefulness may depend on the balance of activity in the GABA/galaninergic systems and orexin/hypercretin systems of the posterior hypothalamus
For introduction to topic see PDF below
- Modulation of Vigilance in the Primary Hypersomnias by Endogenous Enhancement of GABAA Receptors PDF
https://www.researchgate.net/publication/233751072_Modulation_of_Vigilance_in_the_Primary_Hypersomnias_by_Endogenous_Enhancement_of_GABAA_Receptors
- GABAergic system https://bra.in/6joNXY
- GABAergic and galaninergic neurons https://bra.in/4jYbZ2
- Hypocretin (Orexin) Neurons https://bra.in/9j8LNb
- GABA/galaninergic systems https://bra.in/5jk6Bw
- GABA receptors https://bra.in/8qeD3g
- Modulation of Vigilance in the Primary Hypersomnias by Endogenous Enhancement of GABAA Receptors PDF
3c) Ascending Arousal Systems in the Regulation of Sleep and Awakefulness Mediated by the Basal Forebrain
3d) Hypocretin/Orexin System
3e) Reticular Activating System
CFS and Neurology – An overview of recent research Prohealth
Many observations suggest that CFS could derive from residual damage to the reticular activating system (RAS) of the upper brain stem and/or to its cortical projections. It should be pointed out that although the larger right greater than left asymmetry in regional cerebral blood flow is found at the parietotempotal level in CFS patients as compared to healthy controls, no significant correlations are found between frontal tracer uptake and rightleft parietotemporal asymmetry, on the one hand, and clinically relevant CFS dimensions on the other. Damage to the RAS could be produced by a previous viral infection, leaving functional defects unaccompanied by any gross histological changes.
From: Neurophysiology of Sleep and Wakefulness: Basic Science and Clinical Implications
Alertness and associated forebrain and cortical arousal are mediated by several ascending pathways with distinct neuronal components that project from the upper brain stem near the junction of the pons and the midbrain [33]. One pathway innervates the thalamus, and the second extends into the posterior hypothalamus and forebrain. Key cell populations of the ascending arousal pathway include cholinergic, noradrenergic, serotoninergic, dopaminergic, and histaminergic neurons located in the pedunculopontine and laterodorsal tegmental nucleus (PPT/LDT), locus coeruleus, dorsal and median raphe nucleus, and tuberomammillary nucleus (TMN), respectively. Projections from these various cell groups fire in a characteristic pattern to promote arousal. However, every 24 hours the arousal system is inhibited during sleep by sleepactive γaminobutyric acid (GABA)ergic and galaninergic neurons of the ventrolateral preoptic nucleus (VLPO). The interaction between the VLPO and the branches of the ascending arousal pathway is mutually inhibiting, functioning much like an electrical “onoff” switch, enabling the body to maintain a stable state of wakefulness and sleep [34,59,98]. Normally, this “sleepwake switch” design ensures stability between sleep and wakefulness while promoting rapid transitioning between the two behavioral states. Sleep disorders represent a pathology of this switch, which causes individuals to suffer from state instability, with wake intruding into sleep and/or sleep intruding into wake.
waking behavior is indeed maintained by an “ascending reticular activating system,” originating in the upper brainstem adjacent to the junction of the pons and midbrain and continuing on to the diencephalon, where it separates into two branches [72,109]. In fact, it is now known that the ascending arousal system contains two major branches, each comprising discrete cell populations and neurotransmitters [98] (Fig. 11). The first branch innervates the thalamus, activating relay neurons and reticular nuclei essential for thalamocortical transmission. Two cholinergic structures in the brainstem and basal forebrain serve as the origin of these projections to the principal thalamic nuclei – the PPT/LDT nuclei [40]. PPT/LDT neurons are most active during wakefulness and rapid eye movement (REM) sleep and discharge more slowly during nonREM (NREM) sleep, a period when cortical activity is reduced [99]. Transmission to the reticular nucleus of the thalamus is of particular importance, as the site functions as a gating mechanism that can block the generation of thalamocortical rhythms and promote a state of excitability and wakefulness [57]. Other projections from the upper brainstem to the midline and intralaminar thalamic nuclei, which include the reticular formation, the parabrachial nucleus, and the monoaminergic systems (discussed below), are also believed to be involved in cortical arousal [48].
A schematic drawing showing key components of the ascending arousal system. Adapted from Saper 2005, pg 1258 [99].
The second branch of the ascending arousal system projects into the lateral hypothalamus, basal forebrain, and the cerebral cortex [45,96,98]. It comprises a number of monoaminergic cell populations, including noradrenergic neurons of the locus coeruleus, serotoninergic dorsal and median raphe nuclei, dopaminergic neurons of the ventral periaqueductal grey matter, and the histaminergic TMN. Several additional cerebrocortical afferents have been identified: lateral hypothalamic peptidergic neurons, which contain melaninconcentrating hormone or orexin/hypocretin, and basal forebrain nuclei, which contain acetylcholine or GABA [99]. Neurons in these monoaminergic systems have broad action potentials, discharging most rapidly during wakefulness, slowing during NREM sleep, and showing little activity during REM sleep [8,33,110]. A similar pattern was reported in orexin/hypocretin neurons of the lateral hypothalamus [32, 63]. In contrast, melatoninconcentrating neurons, which play an important role in REM homeostasis, are strongly active during REM sleep [119], and cholinergic neurons of the basal forebrain discharge at maximal rates during both REM sleep and active waking [49]. Lesions along this second branch are associated with narcolepsy and other sleep disturbances in rats [37], with the loss of hypocretin cells, in particular, contributing to the difficulty in maintaining arousal and the loss of muscle tone during cataplectic attacks [63].
In sum, cholinergic neurons, monoaminergic cell populations, and orexin/hypocretin nuclei of the lateral hypothalamus located along the two branches of the ascending arousal system, discharge in a stereotypical and coordinated manner to promote cortical arousal, with each making unique, though overlapping and redundant, contributions to achieve and sustain wakefulness. During sleep, these circuits are blocked by neurons of the VLPO.
Following experiments by McGinty and colleagues, which demonstrated that lesions in the basal forebrain suppressed sleep in cats [58], Sherin et al.determined that a group of ventrolateral preoptic neurons is specifically activated during sleep [107]. Neurons of the VLPO form a dense cluster and also extend more diffusely to innervate the monoaminergic systems in the hypothalamus and brainstem that participate in the modulation of cortical arousal (Fig. 22). VLPO efferents contain the inhibitory neurotransmitters GABA and galanin, and have been shown to play a central role in the mammalian brain in quieting the ascending monoaminergic arousal system during sleep [36,106].
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2701283/bin/CN6367_F2.jpg
A schematic drawing showing primary projections of the VLPO to the main components of the ascending arousal system. Adapted from Saper 2005, pg 1258 [99].
Experiments in different animal species have indicated that injury to the VLPO cluster and extended VLPO decreases NREM and REM sleep, respectively [51,53]. Neurons of the extended VLPO connect with pontine sites implicated in REM sleep gating – the LDT, dorsal raphe nucleus, and locus coeruleus [51], whereas the VLPO cluster provides output to histaminergic neurons of the TMN, which, as noted previously, are active during waking, reduce firing during NREM sleep, and cease discharge during REM sleep [42, 50].
Afferents from the components of the monoaminergic arousal system also connect with the VLPO [18]. Noradrenaline and serotonin released by axons from the locus coeruleus and median raphe nuclei, respectively, inhibit VLPO neurons in recordings of cells in hypothalamic slices [34], as do GABA [16] and galanin [46] produced by TMN neurons. (VLPO neurons do not appear to have receptors for histamine.) Thus, the reciprocal inhibitory interaction of sleeppromoting VLPO neurons and the noradrenergic, serotoninergic, and cholinergic waking systems to which they project establishes a remarkable dynamic, in which the VLPO is downregulated by the very arousal systems it blocks during sleep [34,10].
Go to:
THE BRAINSTEM CONTROL OF STATE STABILITY
The reciprocal inhibitory exchange between the major ascending monoaminergic arousal groups and the sleepinducing VLPO acts as a feedback loop; when monoamine nuclei discharge intensively during wakefulness, they inhibit the VLPO, and when VLPO fire rapidly during sleep, block the discharge of the monoamine cell groups [98]. This relationship is described as a bistable, “flipflop” circuit, in which the two halves of the circuit strongly inhibit each other to produce two stable discharge patterns – on or off (Fig. 33). Intermediate states that might be partially “on and off” are resisted. This model helps clarify why sleepwake transitions are relatively abrupt and mammals spend only about 1% to 2% of the day in a transitional state [99]. Hence, changes between sleep and arousal occur infrequently and rapidly. As will be described below, the neural circuitry forming the sleep switch contrasts with homeostatic and circadian inputs, which are continuously and slowly modified [98].
A schematic diagram of the flipflop switch model. Adapted from Saper 2005, pg 1259 [99].
Despite the bistability of the on/off feedback loop, if either side is weakened or injured, unwanted instability can occur during both sleep and wake states, irrespective of which side is damaged. For instance, animals with VLPO lesions experience a 50% to 60% reduction in NREM and REM sleep time and wake up frequently during their sleep cycle [52]. Rapid sleepwake cycling also is common in the elderly [6], who have fewer VLPO neurons [36]. These findings suggest that when the selfreinforcing properties of the circuitry are weakened, individuals shift back and forth between sleep and wakefulness more frequently as well.
Go to:
PROMOTING SLEEPSTATE STABILITY
Orexin (hypocretin) is a neuropeptide produced by the neuronal cluster in the posterior portion of the lateral hypothalamus [24,94]. Neurons containing orexin innervate the major nuclei implicated in sleep regulation. Specifically, orexin1 receptors are found in the locus coeruleus, orexin2 receptors in the TMN, and both types in the median raphe nuclei and mesopontine reticular formation [54]. In contrast, orexin/hypocretin neurons only modestly affect the VLPO [18,95,128]. Therefore, it is possible that orexin/hypocretin may promote wakefulness by upregulating monoaminergic neuronal populations.
This hypothesis has been confirmed in animals and humans. Experiments using orexinreceptor knockout mice produce behavioral symptoms and electroencephalographic results strikingly similar to those of humans with narcolepsy, which is characterized by sleep/wake as well as REM sleep dysregulation [17]. Similarly, humans who have narcolepsy with cataplexy have been shown to have few orexin neurons in the lateral hypothalamus and low orexin levels in the cerebrospinal fluid [61,86,113]. Fos expression in orexin/hypocretin neurons was correlated positively with wakefulness and negatively with NREM and REM sleep [32], whether measured during periods of sleep deprivation or following the use of stimulants such as modafinil [17,101]. Thus, excitatory orexin input may help regulate normal cortical arousal and wakefulness.
Nevertheless, orexin/hypocretindeficient mammals do not exhibit excess sleep. Instead, their onoff circuitry appears unstable, which leads to poor sleepwake maintenance and dysfunctional switching [98]. Mochizuki et al.confirmed that the state instability during wakefulness experienced by orexindeficient mice is not a result of abnormal sleep homeostasis, poor circadian control, or defective monoaminergic systems but rather is the consequence of behavioral state instability due to low betweenstate transitional thresholds [68]. In short, the asymmetric relationship between orexin/hypocretin neurons and arousal and sleeppromoting cell populations may provide stability to the sleepwake system by anchoring the flipflop switch [99].
Go to:
HOMEOSTATIC REGULATION OF SLEEP
Sleep is understood to be restorative, but precisely what is being restored is uncertain. As a homeostatic process, sleep allows the body to return to equilibrium when it is disturbed. For instance, sleep deprivation tends to be followed by extra compensatory sleep to make up for the loss, albeit not on a minuteforminute basis. Borbely and colleagues proposed a twoprocess model of sleep regulation to explain the homeostatic and circadian drives for sleep [1,12]. The homeostatic component, named Process S (sleep), is believed to derive from a substrate or protein that registers a homeostatic “need to sleep” during periods of extended wakefulness that is subsequently relieved during sleep. As NREM sleep appears to take precedence over REM sleep following acute sleep loss, it is probable that the homeostatic mechanisms for the two sleep states differ [99].
The underlying mechanisms remain unclear. However, McCarley and colleagues have shown that adenosine acting in the basal forebrain is a key mediator of homeostatic control (for a more detailed review, see McCarley 2007 [56]). Increased adenosine release accompanies the accumulation of the need to sleep, suggesting that the nucleoside, adenosine, may be involved in the homeostatic control of sleep expression [9]. During periods of wakefulness, glycogen, the body’s principal store of energy, is exhausted [47]. As glycogen is broken down into adenosine, extracellular levels of adenosine begin to accumulate in the basal forebrain [87], leading to the replenishment of glycogen levels with recovered sleep [47,105]. Experimental models showed that the injection of adenosine or an adenosine A1 receptor agonist into the rat basal forebrain or the cat VLPO, respectively, promoted sleep by inhibiting multiple wakepromoting regions of the brain or exciting sleeppromoting cell groups [101,112]. Adenosine also may excite VLPO neurons by disinhibiting GABAergic inputs [16]. Therefore, by inhibiting the basal forebrain arousal system and triggering the VLPO nucleus, adenosine may act as homeostatic regulator of the sleep need. Recent evidence has shown that the sleeppromoting effects of adenosine are further enhanced through its action at the A1 receptor, which triggers an intracellular cascade leading to increased adenosine A1 receptor production [56]. Other mediators of homeostatic drive may be identified in the future.
Go to:
CIRCADIAN SLEEP REGULATION
A second component of the sleepwake regulatory mechanism, which Borbely called Process C, involves circadian influences [1,12]. Dijk and Czeisler confirmed the role of the circadian pacemaker in the timing of the sleepwake cycle and regulation of the internal structure of sleep in a study in which 8 men lived in an environment free of time cues [27]. They found that sleep propensity and sleep structure derive from the interactions of circadian and sleepwakedependent oscillatory processes. The locus of this endogenous circadian pacemaker is the suprachiasmatic nucleus (SCN) of the hypothalamus.
The SCN, which directs the circadian program, has been called the brain’s “master clock” [89]. Circadian timing, in which neurons fire in a 24hour cycle, is organized in a hierarchy of tissuespecific structures located throughout the body. These tissuespecific rhythms are coordinated by the SCN based on light input from the outside world during daytime and by melatonin secretion during the dark cycle [15]. Damage to the SCN eliminates the circadian rhythms of many behaviors, including sleep [69]. In particular, lesions of the retinohypothalamic tract (RHT) of the SCN, which processes light input, cause animals to exhibit freerunning behaviors, demonstrating that the SCN is necessary for synchronization of circadian rhythms to the solar day [44].
Early studies of circadian rhythms in rats with lesions of the SCN suggested that the homeostatic process was independent of the circadian clock, as disrupted circadian sleepwake cycles following SCN destruction were not accompanied by changes in overall sleep duration or recovery after sleep [64,65]. In 1993, however, Edgar and colleagues found that lesions of the primate SCN caused both a loss of circadian timing and increased total sleep time, suggesting an interaction of circadian and homeostatic processes. According to Edgar’s “opponent process” model, the SCN master circadian clock produces an alerting signal to enhance wakefulness and to actively counteract the accumulation of homeostatic drive for sleep [30].
A number of recent experimental studies have clarified the relationship between the SCN and the sleepwake cycle. Most outputs from the SCN are directed at the subparaventricular zone (SPZ) and dorsomedial nucleus of the hypothalamus (DMH); in contrast, there is little innervation of the VLPO or orexin neurons [95,128]. Lu and colleagues showed that, in rats, lesions of the ventral SPZ disrupted measures of the circadian index of sleep by 90% but had little effect on body temperature; whereas, dorsal SPZ lesions lowered the circadian rhythms of temperature by 75% but did not affect sleep (<5%) [53]. This finding indicates that the SPZ is a complex region comprising neuronal subpopulations that differentially regulate circadian rhythms of different physiological responses.
Moreover, the SPZ appears to link circadian input from the SCN to the DMH and preoptic targets, thereby amplifying the circadian responses [53]. The DMH is a particularly important conduit for delivering signals from the SCN to the sleepregulatory system. Lesions of the DMH reduced circadian rhythms of wakefulness, feeding, locomotor activity, and serum corticosteroid levels by 78% to 89% [19]. The DMH also sends GABAergic projections to the sleeppromoting VLPO nucleus and glutamatethyrotropinreleasing hormone afferents to the excitatory lateral hypothalamic area [19]. Therefore, by integrating clock information from the SCN and the SPZ, the DMH plays a major role in regulating circadian sleep behavior.
In sum, an intricate multistage pathway connecting the SCN, the ascending arousal system, and the VLPO via the SPZ and DMH regulates circadian rhythms of sleep and other behaviors (Fig. 44). One rationale for this complexity is that it gives mammals the flexibility to adapt their behavioral and physiological cycles to environmental cues, and therefore establish patterns of restactivity and sleepwakefulness that are best able to meet the organism’s needs [97,99]. For instance, hypothalamic orexin neurons monitor indices of energy balance and mediate adaptive arousal due to food scarcity [127]. Whereas orexin expression in normal mice decreases as levels of blood glucose and leptin rise following food intake, mice with ablated orexin neurons do not respond to fasting with increased arousal and foodseeking activity. Precisely how external forces such as food availability and predation are able to reset homeostatic and circadian systems is currently unknown.
Ascending Arousal Pathway (ARAS)
Ascending Arousal Systems in the Regulation of Sleep and Awakefulness Mediated by the Basal ForebrainRead 'What keeps us awake?
The role of the ascending arousal systems in the
regulation of sleep and wakefulness mediated by
the basal forebrain'
Neuroanatomic Connectivity of the Human Ascending Arousal System Critical to Consciousness and Its Disorders
Fig. (4)
Circadian regulation of sleepwake cycles. Adapted from Fuller 2006, pg 488 [33].
Go to:
THE NEUROPATHOLOGY OF SLEEPWAKE DISORDERS
At present, the pathophysiology of many sleepwake disorders is poorly understood [41]. Generally, a combination of biological, psychological, and social factors is implicated in the etiology of these conditions. The remainder of this review will describe the substrates and mechanisms that have been identified in the most common sleepwake disorders and the clinical implications for the selection of suitable treatment strategies.
Insomnia
Insomnia, the most frequently reported sleep disorder, is characterized as a state of hyperarousal in which stress is believed to activate the hypothalamicpituitaryadrenal axis [41,85]. Vgontzas et al. demonstrated that, compared with healthy subjects, those with chronic insomnia had increased secretion of corticotropin and cortisol throughout the sleepwake cycle [121]. Additionally, Nofzinger and colleagues, using positron emission tomography (PET) studies to assess regional cerebral glucose metabolism, demonstrated that insomnia also is associated with greater wholebrain metabolism during both sleep and wake periods and, notably, a failure of wakepromoting structures to deactivate during the transition from waking to sleep states [78]. Structures regulating the sleepwake cycle, such as the brainstem, hypothalamus, and basal forebrain, are abnormally overactive during sleep. The ventral emotional neural system also is hyperactive during wakefulness in patients with primary insomnia and insomnia associated with depression, and this abnormal activity persists into NREM sleep [77]. These PET findings of whole brain hypermetabolism during sleep and wake states, and reduced waking metabolism in the prefrontal cortex of patients with insomnia, suggest that they have chronic insufficient sleep, which may explain daytime symptoms of fatigue [78]. The results also may explain why cognitive factors (eg, worry) and environmental cues (eg, light exposure and unstable sleep schedules) perpetuate insomnia. A recent review by Roth and colleagues explores the pathophysiology of insomnia and treatment implications in more detail [91]. For more information about treatment for insomnia, see Morin 2005 [71].
Narcolepsy
Narcolepsy is characterized by excessive daytime sleeping and abnormal REM sleep [60,100]. Little is known about the cause of the disorder, and the little that is understood pertains to narcolepsy with cataplexy [23]. As noted above, individuals with this condition have marked neuronal loss (85% to 95%) in the hypothalamic regions responsible for producing orexin/hypocretin, including the dorsal and lateral hypothalamus, as well as in the locus coeruleus, thalamus, and the cerebral cortex [86,113]. The presence of gliosis in the orexin cell region suggests that narcolepsy is the consequence of a neurodegenerative process [113]. Although the precise mechanisms are unknown, it is probable that both genetic and environmental triggers are involved in the onset of the condition [60].
Disorders Associated With Fragmented Sleep
Obstructive sleep apnea (OSA), the most prevalent type of sleep disordered breathing, is characterized by repeated episodes of partial (hypopneas) or complete collapse (apneas) of the pharyngeal airway [41]. The apneic or hypopneic episodes lead to arousal and may induce hypoxemia and hypercapnia, as well as abnormally high levels of sympathetic nervous system activity [74]; these neuropathological changes are mediated by chemoreceptors in the carotid body and brainstem. High levels of sympathetic drive are evident even during periods of wakefulness, when subjects are breathing normally and there is no evidence of chemoreceptor activation [74]. OSA is associated with hypertension, cardiovascular disease, and depression [80,84]. OSA patients have a higher incidence of stroke and transient ischemic attacks as well [76,80].
Another common disorder associated with fragmented sleep is restless legs syndrome (RLS). People with RLS report a strong urge to move the legs, although the arms, trunk, or head and neck may be affected as well [4]. Symptoms of paresthesias worsen at night and at rest, and are relieved by movement, making it difficult to fall asleep or maintain sleep [41]. Altered dopamine and iron metabolism has been proposed as a mechanism of RLS, and a strong familial component has been identified as well [125]. Subjects with RLS have low iron levels in the substantia nigra and putamen (regions of the brain that are responsible for controlling voluntary movement), and this disturbs the normal transmission of dopamine signals [3,116]. More specifically, Connor and colleagues suggested a defect in the regulation of transferrin receptors on neuromelanincontaining cells as a possible trigger [20]. The fact that RLS is common in individuals with iron deficiency provides clinical support for this hypothesis [41]. In addition, dopaminergic agonists have been shown to be effective in the treatment of RLS, whereas dopamine antagonists aggravate sensorimotor symptoms and sleep disturbances [123,124,126]. Taken together, these findings demonstrate that impaired iron metabolism is involved in the pathogenesis of RLS; whether other causative factors are involved is still to be determined [3].
Circadian Rhythm Sleep Disorders
Circadian rhythm sleep disorders stem from a chronic disturbance in the relationship between the circadian pacemaker and environmental cues (eg, the lightdark cycle) [41]. In their 2005 ICSD update, the American Academy of Sleep Disorders identified 9 circadian disorders, including delayed sleep phase type, advanced sleep phase type, shift work type, nonentrained sleepwake type, irregular sleepwake type, and jet lag type [5]. The most prevalent of these conditions is shift work type circadian rhythm sleep disorder, also known as shift work disorder (SWD).
Shift work disorder is characterized by complaints of insomnia or excessive sleepiness (ES) during work hours scheduled during the customary sleep periods [5]; it is estimated to affect about 32% of nightshift workers and 26% of rotatingshift workers [29]. It is believed to be caused by misalignment of circadian regulation and sleepwake behavior (ie, displaced work hours) [2]. Several different kinds of shiftwork schedules may be implicated in the etiology of SWD – night shifts, early morning shifts, and rotating shifts, with the first two predominating. In people with SWD, total sleep time is reduced by 1 to 4 hours, and sleep quality is described as unsatisfactory. Patients may also complain of difficulties with sleep initiation and awakening; ES and diminished alertness, which may impair mental ability and work performance, also are hallmarks of SWD. The ES is due in part to total sleep loss as well as to a decreased circadian alerting signal that corresponds to the altered work hours [5].
The pathology of SWD raises important safety concerns, particularly with respect to the elevated risk of motor vehicle accidents and workplace injury [79]. People with SWD also have higher rates of cardiovascular and gastrointestinal disease and depression, and are more likely to miss family and social activities than are shift workers without the disorder [29]. See Schwartz and Roth 2006 for a more complete review of the burden of illness and management approaches to SWD [104].
Disordered Sleep Associated with Primary Neurological Disorders: Alzheimer’s and Parkinson’s Diseases
Neurodegeneration in brain regions that are involved in sleep regulation can produce sleeppattern abnormalities. Although the pathogenesis of sleep disturbances associated with Alzheimer’s disease (AD) is unknown [21,25], a relationship between disordered sleep and a number of behavioral disturbances associated with dementia, such as aggressiveness and depression, has been identified [70,114]. Individuals with AD also have a higher prevalence of OSA [11], possibly because both conditions are associated with the APOE4 allele.
In contrast, the etiology of Parkinson’s disease (PD)related sleep disorders is better established. (Askenasy 2003 provides a useful introduction [7].) PD is a dopaminergic disease, and patients with PD have fewer dopaminergic neurons in cell groups of the ascending arousal system, relative to controls [129]. Other loci of arousal, such as noradrenergic and cholinergic nuclei, are depleted in PD as well [41]. There also is evidence that Lewy body degeneration in the lower brainstem, substantia nigra, and other mid and forebrain gray matter regions begins relatively early in the disease process [13]. This finding is consistent with the observation that REM sleep disturbances are common in PD and may precede the onset of overt symptoms [13,41]. Despite these insights, much about the neurobiology of disordered sleep secondary to AD, PD, and other primary neurodegenerative conditions remains to be described.
Treatment Considerations
Sleep disturbances are typically treated with a combination of behavioral and pharmacological therapies. The former include various psychological techniques such as cognitive behavioral and sleeprestriction therapy, and strategies to improve sleep hygiene. Pharmacotherapeutic approaches include hypnotic agents to improve sleep onset and maintenance, and wakepromoting agents such as sympathomimetic alerting agents (such as amphetamines and methylphenidate) and modafinil. Other therapies that have been beneficial in primary sleepwake disorders include continuous positive airway pressure (CPAP) for OSA, antidepressants and sodium oxybate for narcolepsy with cataplexy, and benzodiazepine receptor agonists and melatonin selective receptor agonists for insomnia. Dopamine agonists, benzodiazepines, opioids, and anticonvulsants (including gabapentin) are prescribed for RLS; all may be effective in treating various RLSrelated symptoms. The dopamine agonists ropinirole and pramipexole have become firstline agents approved for the treatment of mildtomoderate RLS.
Sleep problems secondary to neurological disease can be more complex. While AD patients with disturbed sleep should have the underlying symptoms addressed in the same manner as patients without dementia, treatment of PDrelated sleep disturbances is complicated by the fact that dopaminergic medications used to treat the primary condition may cause nocturnal wakefulness, decreased shortwave sleep, and decreased sleep continuity at high doses [41]. Paradoxically, dopamine agonists may also cause ES [83], requiring the addition of a wakepromoting medication for symptomatic relief.
Caffeine
Caffeine is often used as the initial treatment for ES caused by sleep deprivation. It achieves its wakepromoting effects by antagonizing adenosinergic neurons located in the hypothalamus and projecting into cells in the cortex, basal forebrain, and reticular activating system [67]. As described previously, endogenous adenosine levels rise as the need for sleep builds [9]. By inhibiting the basal forebrain arousal system and activating the VLPO, adenosine appears to be a fundamental component in the regulation of the homeostatic sleep system. Through its inhibition of A1 adenosine receptors, caffeine prevents sleep onset and maintenance.
It is well established that caffeine affects alertness and cognitive performance, and may reduce the risk of accident and injury in sleepdeprived individuals. Caffeine supplements in doses of 200 mg improved psychomotor performance tasks and vigilance in young, healthy, noncaffeine dependent individuals [73]. However, tolerance can rapidly develop, and there is no longterm evidence validating its use. Caffeine is not indicated for use in narcolepsy, OSA, SWD, or idiopathic hypersomnia.
Sympathomimetic Alerting Drugs
The mechanism of action of sympathomimetic alerting drugs (eg, dextro and methamphetamine, methylphenidate) is direct or indirect stimulation of dopaminergic and noradrenergic nuclei, which in turn heightens the efficacy of the ventral periaqueductal grey area and locus coeruleus, both components of the secondary branch of the ascending arousal system. Amphetamines also activate nonwakepromoting CNS regions, such as the striatum and nucleus accumbens, causing the adverse effects (eg, nervousness, irritability, anorexia, gastrointestinal problems, and rebound hypersomnia), seen with this class of wakepromoting drugs. In addition, amphetamines have a high abuse potential and can lead to dependence. Some patients may also develop tolerance to the alerting effects of these drugs, although the frequency varies across studies [81,82]. For these individuals, changing medications or introducing a drug “holiday” can improve the response [10].
Sympathomimetic drugs have long been used to treat narcolepsy, although many patients find that amphetamines do not provide a sufficient degree of daytime alertness [66]. For these patients, combining 2 or more alerting drugs (eg, sodium oxybate [see below]) can be beneficial. Amphetamines are available in a wide range of formulations and halflives to offer flexibility in dosing. The daily dose range for methylphenidate is up to 60 mg [90]; dextroamphetamine is up to 60 mg; and methamphetamine is 50 mg [38]. Amphetamines carry a relative contraindication for those with preexisting cardiovascular problems, including hypertension. For all patients, regular blood pressure monitoring is recommended.
Antidepressants and Sodium Oxybate as Anticataplectic Drugs
Cataplexy is usually treated with tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs) or norepinephrine reuptake inhibitors (NRIs) [41]. Newer antidepressants that are not TCAs or SSRIs, such as atomoxetine and venlafaxine, also may be effective [10]. In addition, patients may benefit from sodium oxybate. Although the mechanism of sodium oxybate has been studied extensively, it remains unknown [115]. The treatment is especially effective when combined with other alerting drugs and has been shown to reduce nocturnal sleep disruptions and help consolidate sleep [10].
Modafinil and Armodafinil
Modafinil is a unique wakepromoting compound that is pharmacologically and chemically distinct from other CNS stimulants. A substantial body of research has sought to elucidate the precise mechanism of action of modafinil; to date, the question remains unresolved. (See Schwartz 2005 for a detailed review of the pharmacology of the drug [102].) Preclinical studies show that modafinil has low affinity for receptors for noradrenaline, serotonin, GABA, adenosine, or histamine [26,62,108]. However, Scammell and colleagues reported that modafinil increased activity in the TMN and orexin nuclei, 2 regions involved in the promotion of wakefulness [101]. The drug also potentiates noradrenergic nuclei in the VLPO, thereby inhibiting sleep [35]. Consequently, it is believed that by downregulating sleeppromoting neurons in the VLPO, modafinil enables components of the ascending arousal neuronal pathway (eg, the TMN) to remain active [102]. Further research will be necessary to confirm this hypothesis.
Modafinil is approved for the treatment of ES associated with narcolepsy, OSA, and SWD. Multiple randomized clinical trials in these 3 conditions demonstrated that modafinil produced significant improvement, relative to placebo, in subjective and objective measures of ES, including the Epworth Sleepiness Scale, Multiple Sleep Latency Test, and Maintenance of Wakefulness Test [22,28, 117,118]. Overall clinical condition and sustained attention and reaction time, as measured by the Clinical Global Impression of Change and Psychomotor Vigilance Test, respectively, also improved significantly. In addition, patients with SWD given modafinil reported a significantly lower rate of driving accidents or near accidents while commuting [22]. The drug was well tolerated in these clinical trials, with mildtomoderate, transient headache being the primary reported adverse event [22,28,117,118]. There also have been cases of transient insomnia. Overall, modafinil has a superior side effect profile compared with other wakepromoting drugs, and has a low potential for abuse. For these reasons, the agent has become a treatment of choice for narcolepsy, and is the only currently available medication that is approved for OSA patients treated with CPAP with ongoing daytime ES, as well as patients with SWD [102]. The standard dose is 200 to 400 mg daily [88], although some patients with severe sleepiness may require higher doses, and doses up to 600 mg in divided doses have shown a subjective and objective benefit over lower doses in narcolepsy patients (400 mg at 7 am, 200 mg at noon [103].
Armodafinil is the Risomer of modafinil and is approved for treatment of ES associated with OSA, SWD, and narcolepsy. In clinical trials, armodafinil has been shown to improve wakefulness throughout the day in patients with OSA or narcolepsy [41] and during the night shift and the commute home in patients with SWD [30]. The most common adverse events reported in clinical studies of armodafinil were headache, nausea, and insomnia.
Benzodiazepine Receptor and Melatonin Selective Receptor Agonists
Treatment of insomnia typically involves a combination of behavioral and pharmacological approaches. Among the psychological techniques that have demonstrated effectiveness are stimuluscontrol and sleeprestriction therapies, relaxation training, cognitive behavioral therapy, and sleep hygiene education [69]. The most frequently prescribed pharmacotherapies for insomnia are GABA type A (GABAA) modulators. These drugs target the benzodiazepine receptors in the CNS. Benzodiazepine (eg, temazepam, triazolam) and nonbenzodiazepine (zolpidem, eszopiclone) hypnotics may produce daytime sedation side effects in some patients, but these problems do not limit their utility. Benzodiazepine receptor agonists have an abuse and dependence potential (Schedule IV), especially in patients with a history of substance abuse. Careful surveillance of atrisk individuals is recommended. Benzodiazepine hypnotics also may cause nextday cognitive and motor impairments [111,120] and may engender withdrawal symptoms (eg, rebound insomnia, anxiety, irritability, gastrointestinal distress), particularly following longterm or highdose use [39].
The recently approved MT1/MT2 melatonin receptor agonist ramelteon is a proposed alternative to the benzodiazepine receptor agonists for the treatment of insomnia characterized by difficulty with sleep onset [93]. MT1 and MT2 receptors are located in the SCN and contribute to maintenance of the circadian sleepwake cycle. As the drug has no affinity for benzodiazepine, dopamine, and opiate receptors, or for ion channels and receptor transporters, ramelteon has limited potential for abuse and cognitive and functional impairment. A doseranging, doubleblind, crossover study comparing ramelteon, triazolam, and placebo found that, compared with placebo, ramelteon caused no significant effect on these problematic adverse events at up to 20 times the recommended dose (8 mg daily at bedtime) [43]. In contrast, triazolam negatively affected measures of motor and cognitive performance. Ramelteon significantly reduced latency of persistent sleep and increased total sleep time in adults with chronic insomnia in 2 doubleblind randomized, controlled trails [31,92]. There also were no apparent nextday cognitive or motor effects or evidence of rebound insomnia or withdrawal effects following treatment discontinuation in these trials. Ramelteon should be considered for patients with sleeponset insomnia, particularly those who are treatment naïve, who have a history of substance abuse, who are older adults susceptible to the effects of benzodiazepine and nonbenzodiazepine hypnotics, and who require minimal interference with the arousal response.
Go to:
CONCLUSION
Recent insight into the physiological patterns of sleep and wakefulness has shown that different brainprocessing networks and neurochemical systems are involved in both states. The particular neuronal pathways, transmitters, and receptors that make up the ascending arousal system centered in the hypothalamus interact with sleepactive neurons in the VLPO in a flipflop switch to produce distinct sleepwake states with abrupt transitions. Progress in understanding the neural circuitry underlying the regulation of sleepwake states has led to the identification of new mechanisms and substrates, and studies are now underway to investigate these potential targets. In the future, the products of these investigations may offer novel approaches for the treatment of these common and intractable conditions.
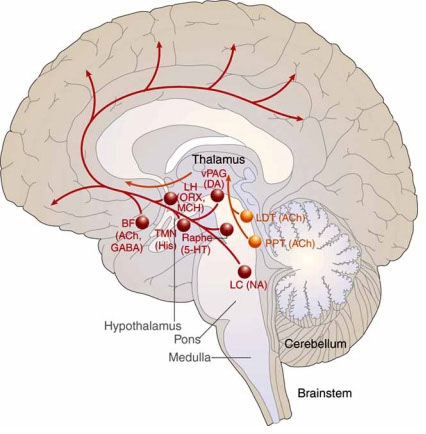
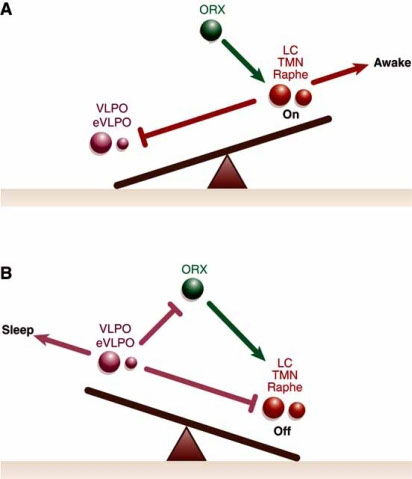
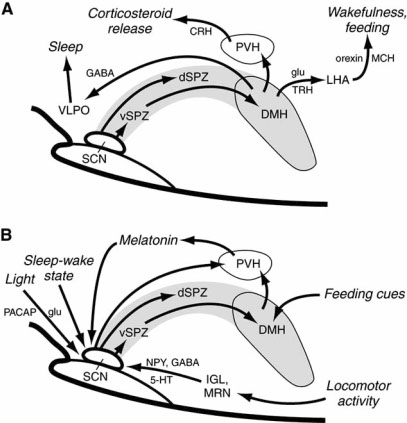
3f) Neuroanatomic Connectivity of the Human Ascending Arousal System Critical to Consciousness and Its Disorders
Neuroanatomic Connectivity of the Human Ascending Arousal System Critical to Consciousness and Its Disorders
From: Neuroanatomic Connectivity of the Human Ascending Arousal System Critical to Consciousness and Its Disorders
The ascending reticular activating system (ARAS) mediates arousal, an essential component of human consciousness. Lesions of the ARAS cause coma, the most severe disorder of consciousness. Because of current methodological limitations, including of postmortem tissue analysis, the neuroanatomic connectivity of the human ARAS is poorly understood. We applied the advanced imaging technique of high angular resolution diffusion imaging (HARDI) to elucidate the structural connectivity of the ARAS in 3 adult human brains, 2 of which were imaged postmortem. HARDI tractography identified the ARAS connectivity previously described in animals and also revealed novel human pathways connecting the brainstem to the thalamus, hypothalamus, and basal forebrain. Each pathway contained different distributions of fiber tracts from known neurotransmitterspecific ARAS nuclei in the brainstem. The histologically guided tractography findings reported here provide initial evidence for humanspecific pathways of the ARAS. The unique composition of neurotransmitterspecific fiber tracts within each ARAS pathway suggests structural specializations that subserve the different functional characteristics of human arousal. This ARAS connectivity analysis provides proof of principle that HARDI tractography may impact the study of human consciousness and its disorders, including in neuropathologic studies of patients dying in coma and the persistent vegetative state.
INTRODUCTION
Human consciousness consists of 2 critical components: arousal and awareness (1, 2). Arousal pathways originating in the brainstem activate awareness networks in the cerebral cortex via synapses in the thalamus and basal forebrain (3–5), or alternatively, via direct innervation of the cortex itself (4, 5). Without arousal, awareness is not possible, as evidenced by comatose patients with brainstem lesions but an anatomically intact cerebral cortex (6, 7). The physiological and neuroanatomic basis of arousal in the brainstem has historically been conceptualized as the ascending reticular activating system (ARAS), an idea introduced by Moruzzi and Magoun in 1949 (8). In classic studies in cats, electrical stimulation of the dorsal midbrain produced widespread, bihemispheric activation of the cerebral cortex, as demonstrated by electroencephalography (EEG). The pathways of the ARAS were initially thought to originate solely in the central core of the upper brainstem called the reticular formation because of its netlike histological appearance (5). In this original model of the ascending arousal system, neural projections from the reticular formation (e.g. the cuneiform/subcuneiform nucleus in the midbrain and pontis oralis in the rostral pons) were believed to activate the cerebral cortex via excitatory (glutamatergic) relays in the thalamus.
It is now recognized that the ARAS is comprised of a complex and diffuse network of neurons projecting from multiple brainstem source nuclei (within and adjacent to the classical reticular core) to the cortex via thalamic (9) and extrathalamic pathways (1, 2, 4, 5, 10). These pathways are typically called “neurotransmitterspecific,” and include serotonergic fibers from the raphe subnuclei of the rostral pons and midbrain (11), noradrenergic fibers from the locus coeruleus of the rostral pons (12), dopaminergic fibers from the ventral tegmental area of the caudal midbrain (13), cholinergic fibers from the pedunculopontine nucleus and laterodorsal tegmental nucleus of the caudal midbrain and rostral pons (3), and glutamatergic fibers from the parabrachial complex in the rostral pons (14). Arousal is further mediated by ARAS connectivity with the hypothalamus, which participates in the regulation of autonomic function (15) and circadian sleepwake cycles (16), and with the basal forebrain, which participates in cortical activation and autonomic integration (5, 14). Thus, in this report “ARAS” refers to the reticular core and extended source nuclei in the brainstem that mediate arousal, as well as their rostral projections to the hypothalamus, thalamus, basal forebrain, and cortex. Of note, these extended source nuclei are the socalled neurotransmitterspecific nuclei. In this modern model of the ARAS, the thalamus is not simply a relay center; rather, it integrates and modulates the interactions between brainstem arousal networks and cortical awareness networks (17).
The elucidation of the neuroanatomic basis of the human ARAS is essential for determining the structural features of human arousal and for treating disorders of consciousness, such as traumatic or strokerelated coma. In addition, postmortem analyses of ARAS connections and their disruption are of major importance for neuropathologists in the elucidation of the neuroanatomic substrate of coma (7), the persistent vegetative state (18), and the minimally conscious state (19) directly in the human brain. Yet, current neuroanatomic models of the human ARAS are based largely upon extrapolations from animal studies, which may not be directly relevant to humans given the unique features of human consciousness. Indeed, it is unknown which pathways in the human ARAS are evolutionarily conserved, and which pathways may have formed new connections during evolutionary development of the human arousal system, as suggested by prior studies showing interspecies differences in brainstem connectivity (20). The critical methodological barrier preventing a detailed connectivity analysis of the human ARAS at autopsy is the limited feasibility of histological tract tracing using postmortem dye injections. This limitation is attributable to excessively long diffusion times (i.e. months) and inability of tracers to diffuse long distances along myelinated axons (21, 22). Moreover, conventional magnetic resonance imaging (MRI) of the brainstem does not provide sufficient resolution to identify the small (i.e. millimeters), discrete components of the ARAS. While functional neuroimaging studies in humans have revealed activation in the brainstem and thalamus during arousal (23), these studies do not provide information about neuroanatomic connectivity between different network nodes. Even tractography reconstructions of diffusion tensor MRI data lack the angular resolution needed to identify the crossing nerve fibers (24) that are a prominent structural feature of the ARAS (5).
Recently, a more sophisticated magnetic resonance technique, high angular resolution diffusion imaging (HARDI) tractography (25), has advanced the study of complex neural networks by enhancing crossing fiber detection (26). Similar to diffusion tensor tractography, HARDI tractography is based on the principle that the neuroanatomic trajectory of axon bundles can be delineated by measuring the directionality of water diffusion along these axons (27, 28). The major methodological advantage of HARDI tractography over diffusion tensor tractography is the ability to resolve multiple axonal bundles traversing in different directions within the same volume of space, or voxel (26). HARDI tractography may, therefore, provide greater spatial and angular resolution for connectivity analyses in the adult human brain than currently available imaging or tissue labeling methods. We hypothesized that HARDI tractography elucidates the complex neuroanatomic connectivity of the human ARAS in the brainstem, hypothalamus, thalamus, and basal forebrain. To test this hypothesis, we performed ARAS connectivity analyses in 2 postmortem human brain specimens, including one in which extensive histological correlation analyses were performed, and in one living human subject. We analyzed the components of the ARAS that originate in the classical reticular core and the extended neurotransmitterspecific nuclei. We refer here to these latter nuclei relative to their known key neurotransmitter according to convention, although we did not perform neurotransmitterspecific immunocytochemical analysis of the postmortem brains. Also of note, we define “connectivity” between neuroanatomic regions by the presence of a bundle of fiber tracts the ends of which terminate within each respective region. This definition of structural connectivity, as delineated by HARDI tractography, does not prove synaptic connectivity, which will require future correlative structuralfunctional studies of the ARAS.
MATERIALS AND METHODS
Human Subjects: Clinical Information and Autopsy Findings
The brains of 3 adults without neurological disease were analyzed with HARDI tractography: 2 were brains from autopsy (Cases 1 and 2), and one was in a living subject (Case 3). Autopsies were performed in Cases 1 and 2 with consent of the family and included permission for research under our institutional reviewboard approved protocol. For Case 3, an in vivo HARDI scan was performed with consent from the subject under a separate institutional reviewboard approved protocol. The brain of Case 1 was used as the index case in which histological data were correlated with the neuroanatomic localization of ARAS nuclei and the connectivity sites of ARAS fiber tracts, as identified by HARDI tractography.
Case 1 was a 53yearold woman with a history of breast cancer (stage I) and a more recent diagnosis of highgrade, pleomorphic sarcoma involving the pelvis. She had no history of neurological illness, brain metastases, or brain radiation. A normal neurological examination was documented during her last hospitalization, 1 month prior to her death in a longterm care facility. The death was attributed to systemic complications of cancer. At autopsy, the fresh brain weight was normal (1,250 g [normal range: 1,200 – 1,500 g]). The leptomeninges showed mild fibrosis. There was mild atherosclerosis of the basilar, middle cerebral, and posterior cerebral arteries, but no occlusions or emboli. The cerebrum and cerebellum showed no macroscopic evidence of atrophy, hemorrhage, or infarcts. Standard sections in a survey of all brain regions demonstrated mild arteriolosclerosis and agonal hypoxicischemic changes in the cerebellar cortex. Microscopic examination of the serial sections of the specimen imaged with HARDI tractography was unremarkable.
Case 2 was a 49yearold man with a history of hypertension, hyperlipidemia, diabetes, and Tcell prolymphocytic leukemia, statuspost unrelated donor stem cell transplantation, who died of sepsis 48 days posttransplant. He had no history of neurological disease or neurological complications of his hematologic malignancy, and a normal neurological examination was documented during his final hospitalization. At autopsy, the fresh brain weight was normal (1,350 g). There was no evidence of atherosclerosis, occlusions, or emboli in the cerebral vasculature; the leptomeninges appeared normal. The cerebrum and cerebellum showed no macroscopic evidence of atrophy, hemorrhage, or infarcts. Standard sections in a survey of all brain regions were stained with hematoxylin and eosin, Luxol fast blue for myelin, and Bodian silver for axons. Microscopic analysis showed mild arteriolosclerosis and scattered reactive astrocytes in gray and white matter. Two microscopic foci of myelin pallor on Luxol fast blue were noted: 1 in the right periventricular white matter and in 1 the cerebellar white matter adjacent to the dentate nucleus, both immediately abutting the ependymal surface. These regions each measured <0.3 cm and showed relative preservation of axons. There were no inflammatory infiltrates. Because of these incidental findings, for which no clinical correlate could be identified on review of all available medical records, an extensive analysis was performed of the brainstem and diencephalic regions that were the focus of our tractography analysis. No additional incidental lesions were identified in the pons, midbrain, hypothalamus, thalamus, or basal forebrain on hematoxylin and eosin or Luxol fast blue stains. Case 3 was a 32yearold man with no history of medical or neurological disease.
Dissection and Imaging of Case 1 Brain Specimen
To scan the ARAS with high spatial and angular resolution, which is critical for tractography analyses of fiber pathways with complex branching patterns and high angles of curvature (24), a small horizontalbore, highfield (4.7 Tesla) Bruker Biospec MRI scanner was used in Case 1. To fit the specimen into the small bore of the scanner, the cerebral hemispheres were dissected from the thalamus and basal forebrain, and the cerebellum was dissected from the brainstem, such that the scanned specimen consisted of the pons, midbrain, hypothalamus, thalamus, basal forebrain, and basal ganglia (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A333). The dimensions of the dissected specimen were 7.0 cm (rostralcaudal axis) × 4.0 cm (anteriorposterior axis at level of thalami) × 5.9 cm (mediallateral axis at level of thalami). The dissection was performed 80 days after the patient’s death and imaging data were acquired 103 days after death.
Immediately prior to scanning, the dissected brain specimen was transferred from a 10% formaldehyde solution to a Fomblin solution (perfluoropolyether, Ausimont USA, Inc., Thorofare, NJ), which reduces magnetic susceptibility artifacts that may occur during image acquisition (29). The HARDI sequence utilized in this scan was a 3dimensional (3D) diffusionweighted spinecho echoplanar imaging (DWSEEPI) sequence with 60 diffusionweighted measurements, corresponding to a cubic lattice in Qspace of b = 4057 s/mm2, using gradient strength of 12.4 G/cm, duration ∂ = 13.4 msec and intertemporal pulse offset ∆ = 25 msec. Repetition time (TR) was 1000 msec, echo time (TE) was 72.5 msec, the field of view was 7.2 × 7.8 × 8.2 cm, and the imaging matrix was 128 × 128 × 128 pixels, yielding a spatial resolution of 562 × 609 × 641 µm. One dataset of 3D DWSEEPI data with b = 0 s/mm2 was also acquired to calculate quantitative diffusion properties in each voxel. Total image acquisition time was 130 minutes.
Imaging of Case 2 Brain Specimen
The brain specimen for Case 2 was scanned as a whole brain on a 3 Tesla Tim Trio MRI scanner (Siemens Medical Solutions, Erlangen, Germany) using a 32channel head coil. As in Case 1, the specimen for Case 2 was transferred from a 10% formaldehyde solution to a Fomblin solution immediately prior to imaging, which was performed 8 months and 16 days after death. To increase diffusion sensitivity on the clinical 3 Tesla MRI scanner that was required to scan a whole brain specimen, diffusion data were acquired using a 3D diffusionweighted steadystate freeprecession (DWSSFP) sequence (30, 31), with a 3DFT readout using following parameters: TR = 27.8 msec, TE = 22.9 msec, flip angle = 60 degrees, bandwidth = 150 Hz/pixel, diffusion gradient duration = 18 msec, diffusion gradient amplitude = 3.2 G/cm, matrix size = 192 × 176 × 128 pixels, and field of view = 19.2 × 17.6 × 12.8 cm, yielding a spatial resolution of 1.0 × 1.0 × 1.0 mm. Four datasets of nondiffusionweighted volumes (b = 0 s/mm2) and 44 datasets of diffusionweighted volumes were acquired, resulting in a total scan time of 5 hours and 35 minutes. Of note, in a DWSSFP sequence, diffusionweighting is not defined by a single global b value, as in the DWSEEPI sequence utilized in Case 1, because the diffusion signal in a DWSSFP sequence cannot be readily dissociated from other imaging properties, such as the T1 relaxation time, the T2 relaxation time, TR, and the flip angle (30, 32). The diffusion weighting in a DWSSFP study is therefore characterized by the diffusion gradient duration and amplitude, as reported above.
Imaging of Case 3
Imaging was performed for Case 3 on a 3 Tesla Tim Trio MRI scanner (Siemens Medical Solutions, Erlangen, Germany) with a 32channel head coil. HARDI data were acquired utilizing a twicerefocused SE EPI sequence (33) with the following parameters: TR/TE = 12750/130 msec, bandwidth = 1395 Hz/pixel, matrix size = 128 × 128, and field of view = 256 × 256 cm2, yielding an inplane spatial resolution of 2.0 × 2.0 mm. Seventyfour slices of 2 mm slice thickness were acquired. Twenty datasets of nondiffusionweighted volumes (b = 0 s/mm2) and 120 datasets of diffusionweighted volumes were acquired with a prespecified b value of 4000 s/mm2, resulting in a total scan time of 29 minutes and 45 seconds.
Histological Methods
At the completion of image acquisition, the dissected brain specimen of Case 1, consisting of the pons, midbrain, hypothalamus, thalamus, basal forebrain, and basal ganglia en bloc (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A333) was divided into 7 blocks, approximately 0.5 to 1.5cmthick that were then embedded in paraffin. Serial transverse sections of the pons and midbrain and coronal sections of the hypothalamus, thalamus, and basal forebrain were prepared at 10µm thickness with a microtome (LEICA RM2255, Leica Microsystems, Buffalo Grove, IL). Every 50th section was stained with hematoxylin and eosin for nuclear boundaries and counterstained with the myelin stain, Luxol fast blue, for fiber tract boundaries. Each examined section was separated by 500 µm, for a total of 74 sections from midpons through the entire hypothalamus, thalamus, basal forebrain, and basal ganglia.
Regions of Interest for ARAS Tractography
ARAS fiber tracts were identified using regions of interest (ROIs) that were manually traced on the diffusion images using TrackVis (version 5.1), an interactive image analysis software program that is available to the scientific community without charge (34). For Case 1, each diffusion image was compared to its corresponding histological section to ensure that radiologic ROIs shared the same borders, size, and contours as the histological ROIs. Histological ROIs were delineated by visual inspection with a light microscope and were confirmed by standard atlases of human neuroanatomy (35–37). ROIs were identified for all of the key ARAS source nuclei implicated in arousal: cuneiform/subcuneiform nucleus, pontis oralis, median and dorsal raphe, locus coeruleus, pedunculopontine nucleus, parabrachial complex (combined medial and lateral parabrachial nuclei), and ventral tegmental area (Fig. 1A, B; Figure, Supplemental Digital Content 2, http://links.lww.com/NEN/A336 and Figure, Supplement Digital Content 3, http://links.lww.com/NEN/A337). ROIs were also traced using histological guidance for the thalamic nuclei implicated in the modulation, or gating, of arousal: the reticular nucleus, the central lateral nucleus, and the centromedian/parafascicular nuclear complex (Figure, Supplemental Digital Content 2, http://links.lww.com/NEN/A336). Each brainstem and thalamic ROI served as a seed point from which fiber tracts were generated. Because the brainstem ROIs are known to change in shape, size and contour along the rostrocaudal axis, histological guidance of ROI tracing was performed for every axial diffusion image using its corresponding histological section. Similarly, each thalamic nucleus was traced on the coronal diffusion images with direct guidance by the location of the stained nuclei on corresponding coronal tissue sections. For the intrathalamic connectivity analyses, a 2ROItractography technique was utilized, based on methods developed by Catani et al (38). Specifically, fiber tracts passing between the reticular nucleus and central lateral nucleus were “virtually dissected” from fiber tracts passing between the reticular nucleus and the centromedian/parafascicular nuclear complex. For Cases 2 and 3, brainstem and thalamic nuclei were traced in accordance with the aforementioned neuroanatomic atlases. We also compared the neuroanatomic localization, contours, and boundaries of these ROIs in Cases 1, 2, and 3 to ensure consistency in the tractography analyses.
Diffusion tractography of the human ascending reticular activating system, as defined by the cuneiform/subcuneiform (CSC) region of interest (ROI). (A, B) Transverse histologic (A) and radiologic (B) sections through the rostral midbrain at the level of superior colliculi (SC) in Case 1. The image in (B) is a nondiffusionweighted image with b = 0 s/mm2; the CSC ROI is labeled in red. Neuroanatomic landmarks: mamillary bodies (MB), cerebral peduncles (CP), red nuclei (RN), superior colliculi (SC) and periaqueductal grey (PAG). (C, D) Ventral (C) and left lateral (D) views of CSC fiber tracts. Tracts are colorcoded according to their direction (red, mediallateral; green, ventraldorsal; blue, rostralcaudal). Bundles of CSC tracts are labeled as follows: VTTR, ventral tegmental tract, rostral; VTTC, ventral tegmental tract, caudal; DTTL, dorsal tegmental tract, lateral; DTTM, dorsal tegmental tract, medial; and MFB, medial forebrain bundle.
There are variations in nomenclature pertaining to the source nuclei of the ARAS in standard neuroanatomic atlases of the human brainstem. In this study, the neuroanatomic localization of the pedunculopontine nucleus was traced according to the brainstem atlas of Paxinos and Huang (35), extending from the caudal midbrain to the caudal border of the red nuclei. The neuroanatomic localization of the parabrachial complex was traced according to the brainstem atlas of Olszewski and Baxter (36), extending from the midpons to the rostral pons.
Tract Construction and Visualization
Diffusion data were processed in Diffusion Toolkit (version 6.0) (34) utilizing a HARDI reconstruction in which the orientation distribution function of angular water diffusion is calculated for each voxel on a spherical harmonic basis (39). By calculating an orientation distribution function for each voxel, the HARDI reconstruction allows for multiple diffusion directions within each voxel, as opposed to the single diffusion direction that is calculated in a diffusion tensor imaging reconstruction. Fiber tracking was performed in TrackVis (version 5.1) (34) utilizing a streamline, deterministic tractography method. Fiber tract propagation is based on the coherence of directional water diffusion in adjacent voxels (40), as determined by the orientation distribution functions generated by the HARDI reconstruction. When the direction of water diffusion is similar in adjacent voxels, the tractography model displays these voxels as being part of a coherent fiber tract. When directional diffusion in multiple voxels is potentially coherent, the tractography algorithm consistently pursues the water diffusion vector of least voxeltovoxel curvature, thereby reducing the possibility of identifying spurious, inaccurate fiber tracts. In this experiment, fiber tracts were terminated when the angle between water diffusion vectors in adjacent voxels exceeded a prespecified threshold of 60°. Whereas tracts tend to terminate at sites of fiber crossing in diffusion tensor tractography, the improved angular resolution provided by HARDI allows tracts to continue in multiple directions at sites of fiber crossing (26). Given that low measurements of directional water diffusion, or fractional anisotropy (FA), have been observed in the dorsal pons, midbrain (41), and thalamus (42) in humans, no FA threshold was applied to the tractography analyses. For intrathalamic connectivity analyses, a length threshold was applied in order to eliminate the long fiber tracts connecting the thalamic nuclei to the brainstem. This length threshold was necessary in order to isolate the comparatively shorter intrathalamic fiber tracts.
To test whether the HARDI data were of sufficient quality for tractography reconstructions (i.e. connectivity modeling), we calculated the signaltonoise ratio for each HARDI scan by dividing the mean signal within the white matter of the internal capsule by the standard deviation of the signal outside the specimen (i.e. noise) on the trace diffusionweighted images and the nondiffusionweighted images with b = 0 s/mm2 (b0). The diffusionweighted images signaltonoise ratio for Cases 1, 2, and 3 were 466, 186, and 137, respectively. The b0 signaltonoise ratio were 49, 106, and 308 respectively, values that are more than sufficient to produce highquality tractography results (25). For the HARDI scans in Cases 1 and 3, we also performed Diffusion Toolkit diffusion tensor reconstructions for standard quantitative measurements of water diffusion – FA and the apparent diffusion coefficient (ADC) – since these measurements have not previously been performed in ARAS fiber tracts. For comparison, FA and ADC were also measured within the corticospinal tract, a structure in which water diffusion has been well characterized in studies of ex vivo (43) and in vivo (44) human brains. The purpose of these comparative FA and ADC analyses was to ascertain how water diffusion in the complex network of ARAS fiber tracts compared to water diffusion within the more homogeneous, parallel fibers of the corticospinal tract. FA and ADC values were not calculated for Case 2 because (as stated above), a single b value cannot be precisely calculated for the DWSSFP sequence that was used in Case 2. As a result, whereas the DWSSFP sequence provides well validated measurements of the directionality of water diffusion for tractography modeling in the human brain (31, 45), the complex signal evolution in the DWSSFP sequence (32) precludes standard quantitative diffusion measurements (31).
To optimize the specificity of ARAS tract identification, we rigorously eliminated nonARAS fiber tracts from the ARAS tractography analysis. NonARAS ROIs that were in close neuroanatomic proximity to ARAS ROIs were first identified and traced on the diffusion images for Case 1 using histological guidance, and then all fiber tracts passing through these nonARAS ROIs were excluded from subsequent ARAS tractography analyses. We eliminated all fiber tracts passing through the superior cerebellar peduncle (both the brachium and the decussation), middle cerebellar peduncle, cranial nerve III, cerebral peduncle (including the frontopontine fibers), medial lemniscus, ventral trigeminothalamic tract (which runs dorsal to the medial lemniscus in the rostral pons), and the medial longitudinal fasciculus (which ascends at the ventromedial margin of the periventricular and periaqueductal grey). Once these nonARAS fiber tracts were eliminated, ARAS connectivity was determined by analyzing the trajectories and termination sites of the ARAS fiber tracts within the brainstem, hypothalamus, thalamus, basal forebrain, and basal ganglia. For Cases 2 and 3, nonARAS pathways were eliminated by tracing nonARAS ROIs using guidance from neuroanatomic atlases. Because the HARDI scans for Cases 2 and 3 were performed on whole brains, we also eliminated fiber tracts from the fornix, a structure that was not present in the dissected specimen of Case 1.
RESULTS
Connectivity of the Cuneiform/Subcuneiform Nucleus and Pontis Oralis
HARDI tractography revealed a bilateral fiber bundle connecting the cuneiform/subcuneiform nucleus in the rostral midbrain to the thalamus, hypothalamus, and basal forebrain (Table 1; Fig. 1). The originating source of this fiber bundle also included the pontis oralis of the rostral pons (Fig. 2). In the rostral midbrain, the bilateral fiber bundle bifurcated into ventral and dorsal bundles (Figs. 1, ,2,2, ,3A).3A). Here we suggest the labels ventral tegmental tract (VTT) and dorsal tegmental tract (DTT) for these 2 large divergent bundles in the human brain, names with historical precedent in experimental animals (46–48). The human VTT bifurcated yet again into 2 additional fiber bundles, one a distinct caudal pathway connecting with the hypothalamus, zona incerta, Forel’s fields, basal forebrain and globus pallidus, and the other a rostral pathway connecting with the paraventricular (midline) region of the thalamus (Table 1; Fig. 3A, 4). We labeled the human hypothalamic VTT pathway as the VTT caudal (VTTC), and the thalamic VTT pathway as the VTT rostral (VTTR).
Diffusion tractography of the cuneiform/subcuneiform nucleus (CSC) and pontis oralis (PO) pathways. (A, B) Left lateral (A) and zoomed left lateral (B) views of the CSC and PO regions of interest, and the fiber tracts that pass through these regions in Case 1. Fiber tracts are colorcoded by the source nucleus of origin (red, CSC; light blue, PO). Tracts in (A) are superimposed on an axial nondiffusionweighted image with b = 0 s/mm2 at the level of the rostral pons. Bundles of CSC and PO fiber tracts are labeled as follows: VTTR, ventral tegmental tract, rostral; VTTC, ventral tegmental tract, caudal; DTTL, dorsal tegmental tract, lateral; DTTM, dorsal tegmental tract, medial; and MFB, medial forebrain bundle. Of note, red fiber tracts generated by the CSC region of interest extend both rostrally and caudally, but the direction of electrical signaling within each fiber tract (i.e. ascending or descending) cannot be determined.
Neuroanatomic connectivity of brainstem arousal pathways and intrathalamic gating pathways. (A) Dorsal view of fiber tracts originating in the pedunculopontine nucleus (purple) and parabrachial complex (yellow) connecting with the centromedian/parafascicular (CEM/Pf) and central lateral (CL) nuclei via the dorsal tegmental tract, medial (DTTM) in Case 1. Pedunculopontine and parabrachial fibers also connect with the reticular nuclei (Ret) via the dorsal tegmental tract, lateral (DTTL), and via an extension of DTTM (arrowheads). Fiber tracts from the dorsal raphe (turquoise), locus coeruleus (dark blue), pedunculopontine nucleus (purple), and parabrachial complex (yellow) connect with the paraventricular, midline nuclei of the thalamus via the ventral tegmental tract, rostral (VTTR). (B) Dorsal view of intrathalamic fiber tracts connecting Ret with CL (red) and Ret with CEM/Pf (pink). Fiber tracts in (A) and (B) are superimposed upon an axial nondiffusionweighted image with b = 0 s/mm2 (b0) at the level of the rostral midbrain and a coronal b0 image at the level of the midthalamus. Axial and coronal images in (A) are semitransparent, so that the trajectories of VTTR fiber tracts are visualized coursing rostrally and dorsally to the paraventricular region of the thalamus. Neuroanatomic landmarks: red nuclei (RN), superior colliculi (SC) and periaqueductal grey (PAG).
pictures at bottom of tab
The pathways that connect each region are the dorsal tegmental tract, medial (DTTM), dorsal tegmental tract, lateral (DTTL), ventral tegmental tract, caudal (VTTC), and ventral tegmental tract, rostral (VTTR).
Neuroanatomy abbreviations: cuneiform/subcuneiform nucleus (CSC), dorsal raphé (DR), Forel’s fields/zona incerta (FF/ZI), intralaminar nuclei of the thalamus (IL), locus coeruleus (LC), lateral geniculate nucleus of the thalamus (LGN), median raphé (MR), paraventricular region of the thalamus (PV), parabrachial complex (PBC), pedunculopontine nucleus (PPN), pontis oralis (PO), pulvinar (Pul), reticular nucleus of the thalamus (Ret), and ventral tegmental area (VTA).
The human DTT bifurcated into 2 additional fiber bundles that each projected to a different distribution of thalamic nuclei. One subdivision, which we labeled the DTT lateral (DTTL), projected laterally to the thalamic reticular nucleus and, to a lesser extent, the basal forebrain, pulvinar, and lateral geniculate nucleus of the thalamus (Table 1; Fig. 1C, D, ,2,2, ,3A,3A, 5). The second subdivision of the DTT, which we labeled DTT medial (DTTM), projected medially to thalamic intralaminar nuclei (central lateral nucleus and centromedian/parafascicular nuclear complex). In addition, several DTTMfibers projected beyond the intralaminar nuclei along a rostral and lateral course to the reticular nucleus (Figs. 3A, 5). Of note, examination of the cuneiform/subcuneiform fiber tracts revealed both short and long rostral projections, and fibers crossing to the contralateral side of the brainstem (Figure, Supplemental Digital Content 4, http://links.lww.com/NEN/A338), wellrecognized features of the brainstem reticular formation (48). In Cases 2 and 3, tractography analyses of ARAS brainstem ROIs revealed VTTR, VTTC, DTTL, and DTTM pathways whose trajectories and sites of connectivity were consistent with the findings in Case 1 (Fig. 5; Figure, Supplemental Digital Content 5, http://links.lww.com/NEN/A339).
Connectivity of the cuneiform/subcuneiform nucleus (CSC) and pontis oralis (PO) in Cases 1, 2, and 3. (A–C) A dorsal view of leftsided CSC fiber tracts (red) and PO fiber tracts (blue) is shown for all 3 high angular resolution diffusion imaging (HARDI) scans (A, Case 1; B, Case 2; C, Case 3). For each scan, a summary of key HARDI sequence parameters is provided below the fiber tracts; the 3 planes (sagittal, coronal, and axial) onto which the fiber tracts are superimposed are shown as nondiffusionweighted images with b = 0 s/mm2. The 2 divergent branches of the dorsal tegmental tract (DTT) – DTT lateral (DTTL) and DTT medial (DTTM) – are seen in each case. In addition, each HARDI scan demonstrates that DTTM connects with and passes through the central lateral nucleus (red, CL) and the centromedian/parafascicular complex (pink, CEM/Pf) of the thalamus, and a bundle of DTTM fiber tracts projects rostrally and laterally to the reticular nucleus of the thalamus (purple, Ret) (white arrows, A, B). Of note, the Ret, CL and CEM/Pf thalamic nuclei are shown in only 1 coronal slice in (B, C) so that the fiber tracts can be optimally visualized, whereas these nuclei are shown as 3dimensional structures in (A). Neuroanatomic landmarks: CC, splenium of the corpus callosum; IC, inferior colliculus; P, pineal gland; Thal, thalamus.
Comparative FA and ADC analyses in the cuneiform/subcuneiform, pontis oralis, and corticospinal fiber tracts demonstrated that FA values were lower and ADC values were higher in the cuneiform/subcuneiform and pontis oralis fiber tracts than in the corticospinal fiber tracts for both Case 1 and Case 3 (Table 2). FA and ADC measurements in the corticospinal fiber tracts in Case 1 were consistent with those reported for ex vivo human brains (43). Of note, the lower ADC values observed in Case 1 as compared to Case 3 are expected because ADC values have been demonstrated to be lower in the postmortem human brain than in the living human brain (43). Interestingly, FA values were similar in Cases 1 and 3, consistent with prior studies showing that tissue death and fixation do not affect FA as much as ADC (43). Whereas there are few prior studies of FA or ADC in living human subjects imaged at high b values, our corticospinal tract FA and ADC measurements are generally consistent with the results of Yoshiura et al, who measured FA and ADC in the internal capsule using the same b value of 4000 s/mm2 that was utilized in Case 3 (49). Also of note, the in vivo white matter ADC values of approximately 400 to 500 × 10−6 mm2/s acquired with a b value of 4000 s/mm2 in this study and in (50) are significantly lower than the ADC values of approximately 700 × 10−6mm2/s that have been reported in prior in vivo studies of human white matter using b values ranging from 700 to 1000 s/mm2 (49, 50). This decline in ADC is explained by the nonmonoexponential relationship between b value and ADC at high b values (where high is typically considered as b > 2000 s/mm2for in vivo human studies) (49, 51).
Table 2pictures at bottom of tab
Fractional Anisotropy and Apparent Diffusion Coefficient Measurements for Cuneiform/Subcuneiform, Pontis Oralis and Corticospinal Fiber Tracts in Cases 1 and 3
FA and ADC values were measured within all fiber tracts projecting from each region of interest. Data are provided as mean ± SD. In the corticospinal fiber tract analysis, tracts were generated using bilateral regions of interest traced on the cerebral peduncles at the level of the red nuclei in the midbrain. To increase the specificity of corticospinal tract analysis, only fiber tracts that also passed through a region of interest traced in the posterior limb of the internal capsule at the level of the midthalamus were included in the analysis. FA values were lower and ADC values were higher in the CSC and PO fiber tracts as compared to the corticospinal fiber tracts, a finding that may be explained by the complex branching of fiber tracts in the CSC and PO pathways, as compared to the parallel composition of fiber tracts in the corticospinal pathway.
ADC, apparent diffusion coefficient; CSC, cuneiform/subcuneiform; FA, fractional anisotropy; PO, pontis oralis.
Connectivity of Monoaminergic, Cholinergic, and GlutamatergicSpecific ARAS Nuclei
Subdivisions of the VTT and DTT were further demonstrated to carry fibers of the known monoaminergic, cholinergic, and glutamatergicrelated source nuclei in the brainstem. Each of these source nuclei had specific patterns of connectivity with the thalamus (Figs. 3A, ,6,6, ,7;7; Figure, Supplemental Digital Content 6, http://links.lww.com/NEN/A341; Figure, Supplemental Digital Content 7, http://links.lww.com/NEN/A342; Figure, Supplemental Digital Content 8, http://links.lww.com/NEN/A343; Figure, Supplemental Digital Content 9, http://links.lww.com/NEN/A344; Figure, Supplemental Digital Content 10, http://links.lww.com/NEN/A345), hypothalamus (Figs. 4, ,6,6, ,7;7; Figure, Supplemental Digital Content 6, http://links.lww.com/NEN/A341; Figure, Supplemental Digital Content 7, http://links.lww.com/NEN/A342; Figure, Supplemental Digital Content 8, http://links.lww.com/NEN/A343; Figure, Supplemental Digital Content 9, http://links.lww.com/NEN/A344; Figure, Supplemental Digital Content 10, http://links.lww.com/NEN/A345), and basal forebrain (Table 1; Figure, Supplemental Digital Content 7, http://links.lww.com/NEN/A342). Of note, diffuse thalamic projections that are known to arise from serotonergic neurons in the raphe and noradrenergic neurons in the locus coeruleus were not identified with ex vivo HARDI tractography in Case 1 but were identified with ex vivo HARDI tractography in Case 2 and in vivo HARDI tractography in Case 3 (Figure, Supplemental Digital Content 11, http://links.lww.com/NEN/A346).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3387430/bin/nihms375366f4.jpg
pictures at bottom of tab
Ascending reticular activating system (ARAS) connectivity with the hypothalamus. (A, B) Ventral (A) and rostral (B) views of ARAS connectivity with the hypothalamus in Case 1. (C) Ventral view from (A) with display of tract endpoints. All fiber tracts and end points are colorcoded according to their source nucleus of origin: turquoise, dorsal raphe; green, median raphe; dark blue, locus coeruleus; purple, pedunculopontine nucleus; yellow, parabrachial complex; and pink, ventral tegmental area. Endpoints for all sourcenucleus tracts terminate in the hypothalamus but only dorsal raphe, locus coeruleus, pedunculopontine, and parabrachial complex pathways connect with the anterior region of the hypothalamus (arrowheads, arrows, and asterisks in A–D. (D) Highzoom view of tract endpoints in the left anterior hypothalamus demonstrates termination sites of locus coeruleus, pedunculopontine, parabrachial complex, and dorsal raphe pathways, some of which overlap within the same voxel (dimensions 562 × 609 × 641 µm). All fiber tracts and endpoints are superimposed upon an axial nondiffusionweighted image with b = 0 s/mm2 (b0) at the level of the red nuclei and a coronal b0 image at the level of the midthalamus (inset). The axial and coronal b0 images in (A) are semitransparent so that the trajectories of fiber tracts are seen as they cross each respective plane. Bundles of fiber tracts are labeled as follows: VTTR, ventral tegmental tract, rostral; VTTC, ventral tegmental tract, caudal. Neuroanatomic landmarks: 3V, third ventricle; CP, cerebral peduncles; RN, red nuclei.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3387430/bin/nihms375366f6.jpg
pictures at bottom of tab
Connectivity of intrathalamic gating pathways and brainstem arousal pathways projecting to the thalamus, hypothalamus, Forel’s fields (FF), and zona incerta (ZI). (A) Dorsal view of intrathalamic fiber tracts that connect the reticular nucleus (Ret) with centromedian/parafascicular complex (CEM/Pf; pink fiber tracts) and that connect Ret with the central lateral nucleus (CL, red fiber tracts) in Case 1. Many red RetCL fiber tracts extend caudally into CEM/Pf, FF, and ZI, and pink RetCEM/Pf tracts also extend into FF and ZI. Brainstem arousal pathways are displayed as fiber tract endpoints, colorcoded by the source nucleus from which the tracts are generated: pedunculopontine nucleus (purple); parabrachial complex (yellow); dorsal raphe (turquoise); locus coeruleus (dark blue); median raphe (green); and ventral tegmental area (pink). (B) Highzoomed dorsal view from within the left CEM/Pf, which is semitransparent to allow for visualization of intrathalamic fiber tracts and pedunculopontine tract endpoints (arrow) within this nuclear complex. (C) Zoomed dorsal view of right side of image in (A) shows that pedunculopontine fiber tracts terminate in FF and ZI (purple end points) in close proximity to fiber tracts that connect these regions to Ret via pathways that pass through CEM/Pf and CL (arrowheads, A, C). Fiber tracts in (A–C) are superimposed on an axial nondiffusionweighted image with b = 0 s/mm2 (b0) at the level of the rostral midbrain and a coronal b0 image at the midthalamus. Neuroanatomic landmarks: 3V, third ventricle, red nuclei (RN), superior colliculi (SC), periaqueductal grey (PAG).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3387430/bin/nihms375366f7.jpg
pictures at bottom of tab
The neuroanatomic connectivity of the serotonergicrelated dorsal raphe (DR). (A) Left lateral oblique view of DR fiber tracts (turquoise) in Case 1 superimposed on an axial nondiffusionweighted image with b = 0 s/mm2 (b0) at the level of the midpons and a coronal b0 image at the level of the midthalamus. The images are semitransparent so that the trajectories of fiber tracts are seen as they cross the coronal plane. (B) Left lateral view superimposed on axial b0 image at the level of the mid pons and a sagittal b0 image at the midline of the specimen. (C) Dorsal view superimposed on an axial b0 image at the level of the inferior colliculi (caudal midbrain) and a coronal b0 image at the dorsal thalamus. Fiber pathways from DR are seen connecting with the inferior colliculi bilaterally (arrows). Bundles of fiber tracts are labeled as follows: VTTR, ventral tegmental tract, rostral; VTTC, ventral tegmental tract, caudal. Neuroanatomic landmarks: red nuclei (RN), superior colliculi (SC) thalamus (Thal), pineal gland (P).
Intrathalamic Connectivity
We delineated connections between the thalamic nuclei implicated in gating of the inputs of the ascending arousal system to the cerebral cortex (17, 52). The reticular nucleus connected with both the central lateral nucleus and the centromedian/parafascicular nuclear complex via dense bundles of fibers with direct, linear trajectories (Fig. 3B). These intrathalamic fiber bundles were interspersed with the DTTM fibers traveling through the intralaminar nuclei to the reticular nucleus (Figure, Supplemental Digital Content 12, http://links.lww.com/NEN/A347). Also of note, a group of fiber tracts was observed to connect the reticular nucleus with Forel’s fields and zona incerta by passing through the central lateral and centromedian/parafascicular nuclei (Figs. 3B, 6). The termination points of these fiber tracts within Forel’s fields and zona incerta were in close proximity to the termination points of brainstem source nuclei pathways that connected with these same regions (Fig. 6).
DISCUSSION
This study provides proof of principle that HARDI tractography is a potent tool for dissecting the complex neuroanatomic substrate of the human ARAS. Virtual dissection of ARAS pathways was demonstrated in the human brain, both in vivo and ex vivo, suggesting that HARDI tractography may be used in living patients and in postmortem neuropathologic analyses to study the neuroanatomic basis of consciousness and its disorders. We found that the human ARAS shares several important features with that of experimental animals: 1) ARAS brainstem source nuclei connect primarily with the intralaminar, paraventricular, and reticular nuclei of the thalamus; 2) the known monoaminergic, cholinergic, and glutamatergicrelated nuclei in the upper brainstem all connect with the hypothalamus; 3) the basal forebrain is connected with multiple brainstem source nuclei of the ARAS; and 4) the reticular, central lateral, and centromedian/parafascicular thalamic nuclei connect with each other, providing a structural basis for human intrinsic thalamic networks that modulate ARAS activation of thalamocortical networks. The short and long rostral projections of the cuneiform/subcuneiform tracts and the fibers crossing to the contralateral side of the brainstem in our 3 cases are also wellrecognized features of the brainstem reticular formation (48) and are consistent with the recognized complexity of the ARAS network (5). Yet, we also defined new human pathways, the VTT and DTT and their subdivisions, which are distinct in their trajectory, connectivity, and source nuclei origins from the VTT and DTT pathways described in animals with tract tracing methods (46–48). In the following discussion, we highlight our findings in the context of the current understanding of ARAS neuroanatomy, with emphasis upon how our data expand upon previous findings in animals and humans. We begin with a consideration of key methodological issues and limitations of HARDI tractography for consideration in the interpretation of our data.
Methodological Considerations and Limitations in HARDI Tractography Analyses
It is important to emphasize that HARDI tractography provides an inferential model, and not a direct measurement, of white matter connectivity in the human brain. In HARDI studies, the accuracy of the neuroanatomic connectivity model depends upon several methodological factors that must be considered in both the data acquisition and postprocessing stages of the experiment. For example, the sensitivity of HARDI tractography for detecting fiber tracts depends primarily on the spatial and angular resolution of the water diffusion measurements that are performed during the scan (data acquisition), which provide the basis for reconstructing a model of white matter connectivity. The specificity of HARDI tractography data is related to the ability to eliminate fiber tracts from nonrelevant neuroanatomic pathways (data postprocessing). In this study, we optimized the sensitivity of ARAS tract identification by acquiring HARDI data with extremely high spatial and angular resolution, particularly in the index case (Case 1), as evidenced by the high number of diffusion gradients (n = 60) (53, 54), the strength of the gradients (12.4 G/cm), which allows for high bvalues with relatively short gradient duration times (55), and the high fieldstrength of the magnet (4.7 Tesla), which allows for high inplane resolution (56). Furthermore, the postmortem, ex vivo data acquisition in Cases 1 and 2 further increases HARDI’s sensitivity for identifying ARAS pathways by allowing for a long duration of scan time (2 hours and 10 minutes for Case 1; 5 hours and 35 minutes for Case 2), as well as the elimination of movement artifacts and susceptibility artifacts caused by the skull base and airfilled sinuses. In Case 3, we increased the sensitivity of in vivo HARDI data acquisition by utilizing a high number of directional diffusion gradients (n = 120) and a high b value (4000 s/mm2). The specificity of ARAS tract identification was optimized during the postprocessing stage by rigorously excluding nonARAS fiber tracts and by defining each ARAS ROI as precisely as possible using histological guidance, as discussed above. However, it should be noted that the ARAS is comprised of multisynaptic pathways that ascend and descend throughout the brainstem; and because HARDI tractography does not provide functional data about the direction of electrical signaling within these pathways, we cannot eliminate the possibility that some of the ARAS pathways identified in this study contained descending fiber tracts. On the other hand, the structural (as opposed to functional) nature of HARDI tractography data enables application of this imaging technique to both in vivo clinical studies and ex vivo neuropathologic studies.
While HARDI and related techniques, such as diffusion spectrum imaging, are better able to resolve crossing fibers than diffusion tensor imaging (24, 26), there is currently a limit at which the spatial and angular resolution, and hence the sensitivity, of these techniques is exceeded. This methodological limitation is particularly relevant for in vivo HARDI studies because of the long duration of scan time that is required to optimize the resolution of HARDI data. We have demonstrated that acquisition of HARDI data with sufficient resolution to identify ARAS connectivity is feasible in a healthy human subject, but human patients with neurological disease may not be able to tolerate longduration HARDI scans (e.g. 30 minutes for Case 3). If HARDI data acquisition parameters are not optimized because of limitations on the duration of scan time, it is possible that small fiber bundles with complex branching patterns may evade detection by HARDI tractography, thereby producing false negative connectivity results. For example, while the VTTR pathway to the paraventricular region of the thalamus was identified by HARDI tractography in all 3 cases in this study, the complex trajectory of this pathway was more clearly delineated in Case 1 than in Cases 2 and 3, likely because of higher spatial resolution of the HARDI data (i.e. smaller voxel size) in the former. Efforts are ongoing to shorten the duration of HARDI data acquisition, improve spatial and angular resolution, and thereby facilitate clinical implementation (57). Yet, even with expected improvements in HARDI data acquisition and postprocessing, HARDI structural connectivity data should be interpreted with caution and should not be considered as proof of functional synaptic connectivity.
Another methodological consideration is that our postprocessing efforts to increase the specificity of ARAS tract identification likely minimized, but did not completely eliminate, the possibility that HARDI tractography could generate spurious tracts, i.e. it may display a connection between 2 fiber bundles that travel through or end within the same voxel but that do not in fact connect (58). False positive connectivity errors that may occur in HARDI tractography experiments may be analogous to leakage of an injected label across neuronal membranes into adjacent, nonrelevant cells in histological tract tracing studies (21). This methodological uncertainty is a particular concern when studying the brainstem components of the ARAS, which arguably contain the greatest concentration of complex crossing fibers of any brain region.
Despite these limitations, HARDI tractography has the ability to generate connectivity maps of complex neural networks in the postmortem human brain more effectively than currently available histological labeling techniques, which are unable to identify long white matter tracts in adults (21). In addition, a significant advantage of HARDI tractography is that it is a noninvasive technique that can be used to investigate white matter connectivity in vivo and ex vivo. Indeed, in vivo tractography data from HARDI and diffusion spectrum imaging studies have been correlated with known neuroanatomic pathways in humans (24, 59), and ex vivo tractography data from these techniques have been correlated with known neuroanatomic pathways in monkeys (24, 60), as well as with histological data in cats (61) and monkeys (60). Furthermore, the feasibility of applying HARDI tractography to postmortem neuropathologic analyses is supported by evidence that the fixation of brain specimens does not alter HARDI data, even when the imaging is performed more than 3 years after death. In a recent diffusion imaging study of 11 human brain specimens that were fixed at an average of 46.2 hours (range 21–69 hours) and imaged at an average of 25.2 months (range 2–40 months) postmortem, the tractography results were consistent with those obtained from in vivo human connectivity studies (43). Although quantitative water diffusion measurements within white matter tracts may be partially dependent on the time to fixation and the time to image acquisition, there was no evidence that these alterations affected the reliability of the postmortem tractography results (43). Importantly, in this study Case 1 was imaged 103 days after death and Case 2 was imaged 8 months and 16 days after death. Thus, imaging of both postmortem brain specimens was performed well within the time window established by Miller et al (2011).
Connectivity of the Cuneiform/Subcuneiform Nucleus and Pontis Oralis
The major finding of our study is that HARDI tractography revealed a bilateral fiber bundle connecting the cuneiform/subcuneiform nucleus in the rostral midbrain to the thalamus, hypothalamus, and basal forebrain, providing a structural basis in the human brain for the physiologic ascending arousal pathway discovered by Moruzzi and Magoun in cats (8). The originating source of this fiber bundle also included the pontis oralis of the rostral pons, consistent with its role in the ARAS based on clinicopathologic studies (6, 7). In the rostral midbrain, the bilateral fiber bundle originating from the mesencephalic and pontine reticular formation, i.e. the reticular core of the classical ARAS, bifurcates into the VTT and DTT, names with historical precedent in experimental animals (46–48). In animal studies, the ventral bundle is mainly a hypothalamic pathway, with divergent projections also to the thalamic reticular nucleus, zona incerta, and Forel’s fields (47, 48). The human VTT, however, bifurcates yet again into 2 additional fiber bundles, one labeled the VTTC, which is a distinct caudal pathway connecting with the hypothalamus, zona incerta, Forel’s fields, basal forebrain and globus pallidus, and the other labeled the VTTR, which is a rostral pathway unexpectedly connecting with the paraventricular (midline) region of the thalamus.
The DTT has been shown in animals to project principally to the thalamic intralaminar and paraventricular nuclei, but not the reticular nucleus of the thalamus (47). In humans, however, we found that the DTT bifurcated into 2 additional fiber bundles that each projected to a different distribution of thalamic nuclei. One subdivision, which we labeled the DTT lateral (DTTL), projected laterally to the thalamic reticular nucleus and, to a lesser extent, the basal forebrain, and the pulvinar and lateral geniculate nucleus in the thalamus. The second subdivision of the DTT, which we labeled DTT medial (DTTM), projected medially to thalamic intralaminar nuclei (central lateral nucleus and centromedian/parafascicular nuclear complex). In addition, several DTTMfibers projected beyond the intralaminar nuclei along a rostral and lateral course to the reticular nucleus.
Thus, the newly defined human VTTR, VTTC, DTTL, and DTTM pathways are distinct in their trajectory, connectivity, and source nuclei origins from the VTT and DTT pathways described in animals with tract tracing methods (46–48). We implicate the DTT and VTT pathways in the mediation of arousal because they connect with thalamic, hypothalamic, and basal forebrain endpoints well established to be crucial to this function. Our structural connectivity findings therefore have significant implications for future functional studies of human arousal, sleepwake cycling, and conscious awareness. Similar to animal studies, this human ARAS analysis suggests a chain of connectivity whereby brainstem source nuclei in the upper pons and midbrain project to the intralaminar nuclei in the thalamus, which in turn project diffusely to the cerebral cortex to activate it (52), thereby enabling cognitive processing. In the human brain, we postulate that the DTTM pathway to the intralaminar nuclei is the main conduit for direct ARAS activation of thalamocortical networks (62). The human DTTL pathway, on the other hand, connects to the thalamic reticular nucleus, which is known to inhibit excitatory input to the cortex via connections with the intralaminar nuclei and which produces the synchronized 7 to 14 Hz oscillations on cortical EEG during the initial stage of sleep (17). Our tractography findings thus suggest a structural basis for the human ARAS to modulate arousal by both direct activation of thalamocortical networks (via DTTM) and by regulation of intrathalamic inhibitory networks (via DTTL).
Connectivity of the Known NeurotransmitterSpecific Components of the ARAS
In this study, the finding that the known neurotransmitterspecific components of the ARAS connect in different patterns with the thalamus, hypothalamus, and basal forebrain underscores the concept that these components underlie different behavioral and/or EEG aspects of the human arousal system. Indeed, each neurotransmitterspecific pathway is believed to become active in anticipation of arousal from sleep, yet with distinct electrophysiological activation patterns (17). The extensive connectivity between known brainstem monoaminergic, cholinergic, and glutamatergicrelated nuclei and the anterior hypothalamus, which modulates circadian sleepwake cycles (16), is consistent with animal data (63) and provides support for current hypotheses that cyclical behavioral states are mediated by reciprocal interactions between interconnected neurotransmitter networks (64). Furthermore, we provide a potential neuroanatomic basis in the human brain for visual imagery in the dream state, as the pathway connecting the pedunculopontine nucleus to the lateral geniculate nucleus (essential to visual processing) is consistent with evidence for cholinergic activation of REM sleep (65).
A potential limitation of HARDI tractography in mapping the connectivity of the human ARAS is related to the observation that our ex vivo connectivity analyses in Case 1 did not identify a wide set of projections to the thalamus from serotonergic and noradrenergicrelated source nuclei. These projections have been suggested by in vivo human neuroimaging studies with serotonergic and noradrenergic ligands (66, 67), and by postmortem neurochemical studies of the thalamus in animals and humans (68–71). We did, however, identify extensive locus coeruleus and dorsal raphe connectivity with the thalamus (particularly with the reticular and centromedian/parafascicular nuclei) in Case 2 (ex vivo) and Case 3 (in vivo), suggesting that HARDI tractography can indeed identify these important noradrenergic and serotonergic pathways in postmortem brain specimens and living subjects.
There are several potential explanations for these locus coeruleus and dorsal raphe connectivity findings, which were unexpected because the spatial resolution in Case 1 was higher than in Cases 2 and 3, and since the angular resolution was similar in Cases 1 and 2. First, HARDI tractography may currently have low sensitivity for identifying tracts with multidirectional branching patterns (72), small axonal diameters (11), and lack of myelination (11)–all known characteristics of the rostral serotonergic and noradrenergic axonal projections. Second, the formaldehyde fixative may interfere with MRI water diffusion measurements along poorly myelinated fiber tracts, although it is unclear why this potential fixative effect would impact the locus coeruleus and dorsal raphe tractography results in Case 1 more than in Case 2 since there was a shorter postmortem interval before imaging in Case 1. Third, while the bvalues used for ex vivo analysis in Case 1 and in vivo analysis in Case 3 were similar (4057 s/mm2 and 4000 s/mm2, respectively), it is possible that at a given b value, the effective diffusion contrast may be greater for in vivoexperimental conditions than for ex vivo experimental conditions because global brain water diffusivity is invariably lower in fixed postmortem tissue. In other words, it may be necessary to use even higher b values (i.e. > 4057 s/mm2) in studies of fixed brain tissue to reach the level of diffusion contrast that can reliably identify the poorly myelinated locus coeruleus and dorsal raphe fiber tracts. Additional studies are therefore needed to determine the optimal HARDI data acquisition parameters (voxel size, number of diffusion gradients, and b value) for identification of each ARAS pathway ex vivo and in vivo. Furthermore, validation of neuroanatomic connectivity models generated by HARDI tractography in ex vivo and in vivo studies ultimately depends upon correlation with anatomic tract tracing studies—either those already available in the literature (47, 60, 73) or, alternatively, in prospective experimental studies designed to confirm unexpected connections. In this regard, the novel ARAS connections that we report here warrant followup in neuroanatomic studies in human tissues and/or animal models.
Intrathalamic Connectivity
The intrathalamic connectivity findings in this study provide a neuroanatomic basis for thalamic modulation, or gating, of the inputs of the ascending arousal system to the cerebral cortex (17, 52). Although we cannot exclude the possibility that some fiber tracts passing between the intralaminar nuclei and the reticular nuclei are thalamocortical projections passing through but not to the reticular nuclei, our finding that the reticular nucleus connects with both the central lateral nucleus and the centromedian/parafascicular nuclear complex is consistent with electrophysiological studies in animals demonstrating specialization of thalamic intralaminar nuclei and the existence of disynaptic intrathalamic pathways mediated by the reticular nucleus (74). Our observation that both the reticular nucleus of the thalamus and the brainstem nuclei of the ARAS connect with Forel’s fields and zona incerta suggests an expanded role for these subthalamic regions as nodes in the neuroanatomic network mediating human arousal, consistent with animal studies (75). Future studies in humans examining ARAS modulation of the activity of the thalamic reticular nucleus may therefore consider the 3 distinct neuroanatomic pathways defined here: 1) the DTTL; 2) the intrathalamic extension of the DTTM; and 3) a potential monosynaptic pathway from the brainstem to Forel’s fields and zona incerta to the reticular nucleus.
In conclusion, the ARAS connectivity results in this study suggest that HARDI tractography has the potential to facilitate the study of the human ascending arousal system in its many physiological manifestations, e.g. sleepwake cycling and anesthesia (76), as well as its pathological conditions such as sleep disorders and coma (6, 7, 77). The multiplicity and redundancy of the ascending arousal system suggest an adaptive mechanism for the recovery of consciousness when one component, but not the full system, is clinically disrupted (6). It remains to be determined which or how many of the multiple components of the ARAS are sufficient for arousal, and hence consciousness, in humans. The examination of HARDI data from multiple patients with different types of ARAS lesions and with different levels of consciousness may ultimately provide us with the answers to such provocative questions as which and how many connections are essential for human cognition. The precise identification of focal disruptions in arousal networks in the brainstem, hypothalamus, thalamus, and basal forebrain of patients with disorders of consciousness may also enable clinicians to target individually tailored pharmacologic (78, 79) and electrophysiologic (e.g. deep brain stimulation (80)), therapies to specific neuroanatomic sites of injury. In addition to its many clinical applications, HARDI tractography can be used in neuropathologic studies to investigate the structural basis for developmental differences in consciousness among the newborn, infant, child, adolescent, and adult, as well as the neuroanatomic basis of coma, vegetative, and minimally conscious states in pediatric and adult patients. We envision that postmortem tractography will be a future adjunct to neuropathologic studies of patients with disorders of consciousness. Specific sites of disruption of ARAS pathways that are not appreciated on gross macroscopic analysis may be identified with postmortem HARDI tractography, and these imaging data may then be used to guide microscopic sectioning, thereby ensuring precise clinicopathologic correlations. The exquisite neuroanatomic delineation of the ARAS by HARDI tractography thus suggests that this advanced imaging technique has the potential to significantly advance the study of the physiology and pathology of human consciousness.

<
>
x
click here to close and return to report











3g) Histaminergic neurons of the hypothalamic tuberomammillary nucleus constitute a major wake‐promoting system
3h) Hypersomnolence, Hypersomnias
Full report https://bra.in/3px4g6 includes:
- Clarithromycin as a Hypersomnia Treatment
- Idiopathic Hypersomnia
- Excessive daytime sleepiness (EDS)
- Histamino‐mimetic drugs such as H3‐receptor inverse agonists could, in fact, become a treatment option not only in patients with narcolepsy (Lin et al., 2008) but also for those with EDS of other origin
- Kleine-Levin Syndrome
- Narcolepsy
- Treatment with Flumazenil (GABAA receptor antagonist)
- Primary Hypersomnia
- Hypersomnolence
- Secondary hypersomnia
Chronic fatigue syndrome (CFS)/myalgic encephalomyelitis (ME) has recently been renamed systemic exertion intolerance disease (SEID). It is similar to sleep disorders such as hypersomnolence (both share the symptoms of excessive daytime sleepiness, non‐restorative sleep, fatigue and cognitive dysfunction). The catagorical/symptomological differences are that CFS/ME/SEID has the additional features of post‐exertional malaise (PEM) and orthostatic intolerance (OI), and that those with hypersomnolence respond differently to stimulants (such as Ritalin and D-Amphetamine).
From: [Bizarre Perpetual Sleepiness Explained](https://www.livescience.com/24984-hypersomnia-sleepiness-treatment.html)
A new treatment may help people with a bizarre medical condition that makes them perpetually sleepy.
The findings, detailed Nov. 21 in the journal Science Translational Medicine, may provide relief for the people who sleep constantly and feel exhausted despite caffeine, other stimulants, and several alarm clocks.
People with [hypersomnia](https://www.livescience.com/9165-sleep-disorder-costly-sufferer-society.html) need to sleep about 70 hours a week and have trouble rousing from sleep. When they are awake, they usually feel as if they've pulled an all-nighter, and describe it as walking around in a fog. Most people come to a diagnosis after conditions like depression, [sleep apnea](https://www.livescience.com/34797-sleep-apnea.html) or thyroid problems have been ruled out, said study co-author David Rye, a sleep researcher at Emory University.
"You look for the typical causes, and then once you rule all those out, you're left with people who still sleep 12, 13, 14, 15 hours," Rye told LiveScience.
For obvious reasons, intense [sleepiness](https://www.livescience.com/5269-study-sleepy.html) can put a crimp in patients' work and social lives. While no one knows exactly how many people have this condition, Rye estimates that roughly 1 in 800 people might be afflicted. Doctors often prescribe patients stimulants such as Ritalin or Adderall, but they don't usually work. [[5 Things You Didn't Know About Sleep](https://www.livescience.com/11386-5-sleep.html)]
So Rye's team wondered whether brain chemicals could cause hypersomnia. Because spinal fluid provides a snapshot of the chemicals floating around the brain, the team took spinal taps from 32 patients with the disease and 16 healthy subjects.
When they put the spinal fluid in a dish with human cells, nothing happened.
So the researchers added a chemical called gamma-aminobutyric acid (GABA), which helps the body shut down. The spinal fluid of the ultra-sleepy amplified the [effects of GABA](https://www.livescience.com/21653-brain-chemicals-sleep-paralysis.html), making it bind much more often to the human cells. (Past research showed a link between GABA and [sleep paralysis](https://www.livescience.com/5646-terrifying-sleep-paralysis-attention.html), or the phenomenon in which one wakes up while his or her muscles are still frozen.)
The sleepy patients were producing a brain chemical that kept them half-sedated all of the time, Rye said.
In a petri dish, adding a drug called flumenazil, which revives patients who have overdosed on sedatives such as Valium, reversed the effects of the sleepy peoples' spinal fluid.
They then tested flumenazil in the sleepy patients. Before taking the drug, hypersomniacs performed as well as the extremely sleep-deprived or slightly inebriated on a test of alertness.
"They're walking around essentially legally drunk all day," he said.
Afterwards, the sleepy cohort performed almost as well as healthy individuals.
The findings suggest that the drug could be an effective treatment for those with hypersomnia, but a follow-up study needs to prove that they actually sleep less at night, he said.
Currently, the drug is only used to treat drug overdoses or to awaken patients unconscious from anesthesia, so the amount of the drug currently produced could only treat a handful of patients. Production would need to increase before it could be widely used, he said.
From: [Hypersomnia - Wikipedia](https://en.wikipedia.org/wiki/Hypersomnia)
Hypersomnia, or hypersomnolence, is a [neurological disorder](https://en.wikipedia.org/wiki/Neurological_disorder "Neurological disorder") of excessive time spent sleeping or [excessive sleepiness](https://en.wikipedia.org/wiki/Excessive_daytime_sleepiness "Excessive daytime sleepiness"). It can have many possible causes^[1]^ and can cause distress and problems with functioning.^[2]^ In the fifth edition of the *[Diagnostic and Statistical Manual of Mental Disorders](https://en.wikipedia.org/wiki/Diagnostic_and_Statistical_Manual_of_Mental_Disorders "Diagnostic and Statistical Manual of Mental Disorders")*, ([DSM-5](https://en.wikipedia.org/wiki/DSM-5 "DSM-5")), hypersomnolence, of which there are several subtypes, appears under [sleep-wake disorders](https://en.wikipedia.org/wiki/Sleep-wake_disorder "Sleep-wake disorder").^[3]^
Symptoms[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=1 "Edit section: Symptoms")]
The main symptom of hypersomnia is [excessive daytime sleepiness](https://en.wikipedia.org/wiki/Excessive_daytime_sleepiness "Excessive daytime sleepiness") (EDS), or prolonged nighttime sleep,^[4]^ which has occurred for at least 3 months prior to diagnosis.^[5]^
Epidemiology[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=2 "Edit section: Epidemiology")]
Hypersomnia affects approximately 5% of the general population,^[6]^ "with a higher prevalence for men due to the [sleep apnea](https://en.wikipedia.org/wiki/Sleep_apnea "Sleep apnea") syndromes".^[5]^
Diagnosis[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=3 "Edit section: Diagnosis")]
"The severity of daytime sleepiness needs to be quantified by subjective scales (at least the [Epworth Sleepiness Scale](https://en.wikipedia.org/wiki/Epworth_Sleepiness_Scale "Epworth Sleepiness Scale")) and objective tests such as the [multiple sleep latency test](https://en.wikipedia.org/wiki/Multiple_sleep_latency_test "Multiple sleep latency test") (MSLT)."^[5]^ The Stanford sleepiness scale (SSS) is another frequently-used subjective measurement of sleepiness.^[7]^After it is determined that EDS is present, a complete medical examination and full evaluation of potential disorders in the differential diagnosis (which can be tedious, expensive and time-consuming) should be undertaken.^[5]^
Differential diagnosis[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=4 "Edit section: Differential diagnosis")]
Hypersomnia can be primary (of central/brain origin), or it can be secondary to any of numerous medical conditions. More than one type of hypersomnia can coexist in a single patient. Even in the presence of a known cause of hypersomnia, the contribution of this cause to the complaint of EDS needs to be assessed. When specific treatments of the known condition do not fully suppress EDS, additional causes of hypersomnia should be sought.^[8]^ For example, if a patient with sleep apnea is treated with CPAP ([continuous positive airway pressure](https://en.wikipedia.org/wiki/Continuous_positive_airway_pressure "Continuous positive airway pressure")) which resolves their apneas but not their EDS, it is necessary to seek other causes for the EDS. [Obstructive sleep apnea](https://en.wikipedia.org/wiki/Obstructive_sleep_apnea "Obstructive sleep apnea") “occurs frequently in [narcolepsy](https://en.wikipedia.org/wiki/Narcolepsy "Narcolepsy") and may delay the diagnosis of narcolepsy by several years and interfere with its proper management.”^[9]^
Primary hypersomnias[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=5 "Edit section: Primary hypersomnias")]
The true primary hypersomnias include these: [narcolepsy](https://en.wikipedia.org/wiki/Narcolepsy "Narcolepsy") (with and without [cataplexy](https://en.wikipedia.org/wiki/Cataplexy "Cataplexy")); [idiopathic hypersomnia](https://en.wikipedia.org/wiki/Idiopathic_hypersomnia "Idiopathic hypersomnia"); and recurrent hypersomnias (like [Klein-Levin syndrome](https://en.wikipedia.org/wiki/Klein-Levin_syndrome "Klein-Levin syndrome")).^[5]^
Primary hypersomnia mimics[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=6 "Edit section: Primary hypersomnia mimics")]
There are also several genetic disorders that may be associated with primary/central hypersomnia. These include the following: [Prader-Willi syndrome](https://en.wikipedia.org/wiki/Prader-Willi_syndrome "Prader-Willi syndrome"); [Norrie disease](https://en.wikipedia.org/wiki/Norrie_disease "Norrie disease"); [Niemann–Pick disease, type C](https://en.wikipedia.org/wiki/Niemann%E2%80%93Pick_disease,_type_C "Niemann–Pick disease, type C"); and [myotonic dystrophy](https://en.wikipedia.org/wiki/Myotonic_dystrophy "Myotonic dystrophy"). However, hypersomnia in these syndromes may also be associated with other secondary causes, so it is important to complete a full evaluation. Myotonic dystrophy is often associated with SOREMPs ([sleep onset REM periods](https://en.wikipedia.org/wiki/Sleep_onset "Sleep onset"), such as occur in narcolepsy).^[5]^
There are many [neurological disorders](https://en.wikipedia.org/wiki/Neurological_disorders "Neurological disorders") that may mimic the primary hypersomnias, narcolepsy and idiopathic hypersomnia: [brain tumors](https://en.wikipedia.org/wiki/Brain_tumors "Brain tumors"); [stroke-provoking lesions](https://en.wikipedia.org/wiki/Stroke "Stroke"); and dysfunction in the [thalamus](https://en.wikipedia.org/wiki/Thalamus "Thalamus"), [hypothalamus](https://en.wikipedia.org/wiki/Hypothalamus "Hypothalamus"), or [brainstem](https://en.wikipedia.org/wiki/Brainstem "Brainstem"). Also, neurodegenerative conditions such as [Alzheimer's disease](https://en.wikipedia.org/wiki/Alzheimer%27s_disease "Alzheimer's disease"), [Parkinson's disease](https://en.wikipedia.org/wiki/Parkinson%27s_disease "Parkinson's disease"), or [multiple system atrophy](https://en.wikipedia.org/wiki/Multiple_system_atrophy "Multiple system atrophy") are frequently associated with primary hypersomnia. However, in these cases, one must still rule out other secondary causes.^[5]^
Early [hydrocephalus](https://en.wikipedia.org/wiki/Hydrocephalus "Hydrocephalus") can also cause severe EDS.^[10]^ Additionally, [head trauma](https://en.wikipedia.org/wiki/Head_trauma "Head trauma") can be associated with a primary/central hypersomnia, and symptoms similar to those of idiopathic hypersomnia can be seen within 6–18 months following the trauma. However, the associated symptoms of headaches, memory loss, and lack of concentration may be more frequent in head trauma than in idiopathic hypersomnia. "The possibility of secondary narcolepsy following head injury in previously asymptomatic individuals has also been reported."^[5]^
Secondary hypersomnias[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=7 "Edit section: Secondary hypersomnias")]
Secondary hypersomnias are extremely numerous.
Hypersomnia can be secondary to disorders such as [clinical depression](https://en.wikipedia.org/wiki/Clinical_depression "Clinical depression"), [multiple sclerosis](https://en.wikipedia.org/wiki/Multiple_sclerosis "Multiple sclerosis"), [encephalitis](https://en.wikipedia.org/wiki/Encephalitis "Encephalitis"), [epilepsy](https://en.wikipedia.org/wiki/Epilepsy "Epilepsy"), or [obesity](https://en.wikipedia.org/wiki/Obesity "Obesity").^[11]^ Hypersomnia can also be a symptom of other sleep disorders, like [sleep apnea](https://en.wikipedia.org/wiki/Sleep_apnea "Sleep apnea").^[11]^ It may occur as an [adverse effect](https://en.wikipedia.org/wiki/Adverse_effect "Adverse effect") of taking certain medications, of withdrawal from some medications, or of drug or alcohol abuse.^[11]^ A genetic predisposition may also be a factor.^[11]^ In some cases it results from a physical problem, such as a tumor, head trauma, or dysfunction of the [autonomic](https://en.wikipedia.org/wiki/Autonomic_nervous_system "Autonomic nervous system") or [central nervous system](https://en.wikipedia.org/wiki/Central_nervous_system "Central nervous system").^[11]^
Sleep apnea is the second most frequent cause of secondary hypersomnia, affecting up to 4% of middle-aged adults, mostly men. [Upper airway resistance syndrome](https://en.wikipedia.org/wiki/Upper_airway_resistance_syndrome "Upper airway resistance syndrome") (UARS) is a clinical variant of sleep apnea that can also cause hypersomnia.^[5]^ Just as other sleep disorders (like narcolepsy) can coexist with sleep apnea, the same is true for UARS. There are many cases of UARS in which EDS persists after CPAP treatment, indicating an additional cause, or causes, of the hypersomnia and requiring further evaluation.^[8]^
Sleep movement disorders, such as [restless legs syndrome](https://en.wikipedia.org/wiki/Restless_legs_syndrome "Restless legs syndrome") (RLS) and [periodic limb movement disorder](https://en.wikipedia.org/wiki/Periodic_limb_movement_disorder "Periodic limb movement disorder") (PLMD or PLMS) can also cause secondary hypersomnia. Although RLS does commonly cause EDS, PLMS does not. There is no evidence that PLMS plays "a role in the etiology of daytime sleepiness. In fact, two studies showed no correlation between PLMS and objective measures of EDS. In addition, EDS in these patients is best treated with psychostimulants and not with dopaminergic agents known to suppress PLMS."^[8]^
[Neuromuscular diseases](https://en.wikipedia.org/wiki/Neuromuscular_disease "Neuromuscular disease") and [spinal cord diseases](https://en.wikipedia.org/wiki/Spinal_cord_diseases "Spinal cord diseases") often lead to sleep disturbances due to respiratory dysfunction causing sleep apnea, and they may also cause [insomnia](https://en.wikipedia.org/wiki/Insomnia "Insomnia") related to pain.^[12]^ "Other sleep alterations, such as periodic limb movement disorders in patients with spinal cord disease, have also been uncovered with the widespread use of [polysomnography](https://en.wikipedia.org/wiki/Polysomnography "Polysomnography")."^[12]^
Primary hypersomnia in [diabetes](https://en.wikipedia.org/wiki/Diabetes "Diabetes"), [hepatic encephalopathy](https://en.wikipedia.org/wiki/Hepatic_encephalopathy "Hepatic encephalopathy"), and [acromegaly](https://en.wikipedia.org/wiki/Acromegaly "Acromegaly") is rarely reported, but these medical conditions may also be associated with the secondary hypersomnias [sleep apnea](https://en.wikipedia.org/wiki/Sleep_apnea "Sleep apnea") and [periodic limb movement disorder](https://en.wikipedia.org/wiki/Periodic_limb_movement_disorder "Periodic limb movement disorder") (PLMD).^[5]^
[Chronic fatigue syndrome](https://en.wikipedia.org/wiki/Chronic_fatigue_syndrome "Chronic fatigue syndrome") and [fibromyalgia](https://en.wikipedia.org/wiki/Fibromyalgia "Fibromyalgia") can also be associated with hypersomnia. Regarding chronic fatigue syndrome, it is "characterized by persistent or relapsing fatigue that does not resolve with sleep or rest. Polysomnography shows reduced sleep efficiency and may include [alpha intrusion](https://en.wikipedia.org/wiki/Alpha_intrusion "Alpha intrusion") into sleep [EEG](https://en.wikipedia.org/wiki/EEG "EEG"). It is likely that a number of cases labeled as chronic fatigue syndrome are unrecognized cases of upper airway resistance syndrome"^[13]^ or other sleep disorders, such as narcolepsy, sleep apnea, PLMD, etc.^[14]^
Similarly to chronic fatigue syndrome, [fibromyalgia](https://en.wikipedia.org/wiki/Fibromyalgia "Fibromyalgia") also may be associated with anomalous alpha wave activity (typically associated with arousal states) during [NREM](https://en.wikipedia.org/wiki/NREM "NREM") sleep.^[15]^ Also, researchers have shown that disrupting stage IV sleep consistently in young, healthy subjects causes a significant increase in muscle tenderness similar to that experienced in "neurasthenic musculoskeletal pain syndrome". This pain resolved when the subjects were able to resume their normal sleep patterns.^[16]^
[Chronic kidney disease](https://en.wikipedia.org/wiki/Chronic_kidney_disease "Chronic kidney disease") is commonly associated with sleep symptoms and excessive daytime sleepiness. For those on [dialysis](https://en.wikipedia.org/wiki/Dialysis "Dialysis"), approximately 80% have sleep disturbances. Sleep apnea can occur 10 times as often in [uremic](https://en.wikipedia.org/wiki/Uremia "Uremia") patients than in the general population and can affect up to 30-80% of patients on dialysis, though nighttime dialysis can improve this. About 50% of dialysis patients have hypersomnia, as severe kidney disease can cause uremic encephalopathy, increased sleep-inducing [cytokines](https://en.wikipedia.org/wiki/Cytokines "Cytokines"), and impaired sleep efficiency. About 70% of dialysis patients are affected by insomnia, and RLS and PLMD affect 30%, though these may improve after dialysis or kidney transplant.^[17]^
Most forms of cancer and their therapies can cause fatigue and disturbed sleep, affecting 25-99% of patients and often lasting for years after treatment completion. "Insomnia is common and a predictor of fatigue in cancer patients, and polysomnography demonstrates reduced sleep efficiency, prolonged initial sleep latency, and increased wake time during the night." [Paraneoplastic syndromes](https://en.wikipedia.org/wiki/Paraneoplastic_syndromes "Paraneoplastic syndromes") can also cause insomnia, hypersomnia, and [parasomnias](https://en.wikipedia.org/wiki/Parasomnia "Parasomnia").^[17]^
[Autoimmune diseases](https://en.wikipedia.org/wiki/Autoimmune_disease "Autoimmune disease"), especially [lupus](https://en.wikipedia.org/wiki/Lupus "Lupus") and [rheumatoid arthritis](https://en.wikipedia.org/wiki/Rheumatoid_arthritis "Rheumatoid arthritis") are often associated with hypersomnia, as well. [Morvan's syndrome](https://en.wikipedia.org/wiki/Morvan%27s_syndrome "Morvan's syndrome") is an example of a more rare autoimmune illness that can also lead to hypersomnia.^[17]^ [Celiac disease](https://en.wikipedia.org/wiki/Celiac_disease "Celiac disease") is another autoimmune disease associated with poor sleep quality (which may lead to hypersomnia), "not only at diagnosis but also during treatment with a gluten-free diet."^[18]^ There are also some case reports of central hypersomnia in celiac disease.^[19]^ And RLS "has been shown to be frequent in celiac disease," presumably due to its associated iron deficiency.^[18]^^[19]^
[Hypothyroidism](https://en.wikipedia.org/wiki/Hypothyroidism "Hypothyroidism") and [iron deficiency](https://en.wikipedia.org/wiki/Iron_deficiency "Iron deficiency") with or without (iron-deficiency [anemia](https://en.wikipedia.org/wiki/Anemia "Anemia")) can also cause secondary hypersomnia. Various tests for these disorders are done so they can be treated.^[20]^ Hypersomnia can also develop within months after viral infections such as [Whipple's disease](https://en.wikipedia.org/wiki/Whipple%27s_disease "Whipple's disease"), [mononucleosis](https://en.wikipedia.org/wiki/Mononucleosis "Mononucleosis"), [HIV](https://en.wikipedia.org/wiki/HIV "HIV"), and [Guillain–Barré syndrome](https://en.wikipedia.org/wiki/Guillain%E2%80%93Barr%C3%A9_syndrome "Guillain–Barré syndrome").^[5]^
Behaviorally induced insufficient sleep syndrome must also be considered in the differential diagnosis of secondary hypersomnia. This disorder occurs in individuals who fail to get sufficient sleep for at least three months. In this case, the patient has [chronic sleep deprivation](https://en.wikipedia.org/wiki/Chronic_sleep_deprivation "Chronic sleep deprivation") although he or she is not necessarily aware of it. This situation is becoming more prevalent in western society due to the modern demands and expectations placed upon the individual.^[5]^
Many [medications](https://en.wikipedia.org/wiki/Medication "Medication") can also lead to secondary hypersomnia. Therefore, a patient's complete medication list should be carefully reviewed for sleepiness or fatigue as side effects. In these cases, careful withdrawal from the possibly offending medication(s) is needed; then, medication substitution can be undertaken.^[5]^
[Mood disorders](https://en.wikipedia.org/wiki/Mood_disorders "Mood disorders"), like depression, [anxiety disorder](https://en.wikipedia.org/wiki/Anxiety_disorder "Anxiety disorder") and [bipolar disorder](https://en.wikipedia.org/wiki/Bipolar_disorder "Bipolar disorder"), can also be associated with hypersomnia. The complaint of EDS in these conditions is often associated with poor sleep at night. "In that sense, insomnia and EDS are frequently associated, especially in cases of depression."^[5]^ Hypersomnia in mood disorders seems to be primarily related to "lack of interest and decreased energy inherent in the depressed condition rather than an increase in sleep or REM sleep propensity". In all cases with these mood disorders, the MSLT is normal (not too short and no SOREMPs).^[5]^
Treatment[[edit](https://en.wikipedia.org/w/index.php?title=Hypersomnia&action=edit§ion=8 "Edit section: Treatment")]
Although "there has been no cure of chronic hypersomnia", there are several treatments that may improve patients' quality of life, depending on the specific cause or causes of hypersomnia that are diagnosed.^[5]^
From: [A review of sleep disturbances following traumatic brain injury | Sleep Science and Practice | Full Text](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4)
Increased sleep need is a significant issue in the acute period following TBI (Baumann et al. [2007](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR7); Sommerauer et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR68); Raikes and Schaefer [2016](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR63)). A prospective study of 96 patients with TBI demonstrated that 22% experienced hypersomnia following TBI, defined as a sleep need of equal to or greater than 2 h when compared to pre-TBI sleep need (Baumann et al. [2007](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR7)). Although no correlations were noted with regards to cerebrospinal fluid (CSF) hypocretin levels, polysomnography (PSG) or multiple sleep latency tests (MSLT), post-TBI patients reporting hypersomnia suffered more severe TBI than those without (Baumann et al. [2007](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR7)). A retrospective case-control study (*n* = 36) showed that patients with hypersomnia based initially on actigraphy testing demonstrated increased stage 3 sleep on subsequent PSG testing when compared to controls (Sommerauer et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR68)).
While it is clear that hypersomnia affects a significant number of patients following TBI, the length of time that this persists is variable. A recent prospective study (*n* = 17) used actigraphy to demonstrate that an increased sleep need might be seen in the acute period following TBI, resolving one month post-injury (Raikes and Schaefer [2016](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR63)). A larger prospective study (*n* = 748) showed that these changes persist up to one month, but may resolve by one year following TBI; however, increased sleep need was assessed via survey rather than actigraphy (Watson et al. [2007](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR75)). Finally, a case-control study evaluating 42 patients with first-time TBI showed that sleep need was still significantly increased at 6 months when compared to controls (Imbach et al. [2015](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR29)). The development of hypersomnia following TBI is a significant predictor of negative social outcomes, including subjective difficulties for patients at work, in relationships and various social settings (Chan and Feinstein [2015](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR14)). This highlights the need for early assessment and treatment of hypersomnia.
Insomnia due to post-traumatic headache
Headache following TBI is a common symptom seen in 20–46.8% of patients with TBI (*n* = 443) (Lavigne et al. [2015](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR37); Chaput et al. [2009](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR15)). This symptom can have a significant impact on quality of life both during wakefulness and sleep and can be seen irrespective of the severity of the injury. Multiple studies have been performed to evaluate the impact of post-traumatic headache (PTH) on sleep, with insomnia the most common symptom experienced (Minen et al. [2016](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR48); Hou et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR27)). A retrospective cohort study (*n* = 98) showed that headache and insomnia are frequently comorbid conditions in the mild TBI population, with up to half of patients with PTH also suffering from insomnia (Hou et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR27)). This study also showed that PTH portended a higher risk of development of insomnia when compared to severity of TBI, with estimates from 12.5 to 27% (Hou et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR27); Jaramillo et al. [2016](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR32)). Finally, multiple studies have shown that insomnia appears to predict the persistence of PTH in TBI patients, leading to a vicious cycle where each symptom promotes the presence of the other. (Chaput et al. [2009](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR15); Hou et al. [2013](https://sleep.biomedcentral.com/articles/10.1186/s41606-018-0020-4#CR27)).
3i) Fatigue (customary or chronic)
TBI and hormones
Biological Processes Associated
With FatigueDiseases associated with fatigue
(Disease Hierarchy)
Traumatic Brain Injury, Panhypopituitarism and Hormonal Evaluations as a Standard of Care
http://www.nevadaosteopathic.org/attachments/article/33/Clearfield%20Panhypopituitarism%20and%20TBI.pdf
Constant fatigue is the #1 symptom across TBI
100% of TBI patients experience fatigue. From there, 68% reported fatigue at 2 years post-injury, at 5 years post-injury 73% reported problems with fatigue.
Page: /
Traumatic Brain Injury, Panhypopituitarism and Hormonal Evaluations as a Standard of Care
http://www.nevadaosteopathic.org/attachments/article/33/Clearfield%20Panhypopituitarism%20and%20TBI.pdf
Constant fatigue is the #1 symptom across TBI
100% of TBI patients experience fatigue. From there, 68% reported fatigue at 2 years post-injury, at 5 years post-injury 73% reported problems with fatigue.
Fatigue from TBI and Factors contributing to chronic fatigue after traumatic brain injury
From: Long-Lasting Mental Fatigue After Traumatic Brain Injury – A Major Problem Most Often Neglected Diagnostic Criteria, Assessment, Relation to Emotional and Cognitive Problems, Cellular Background, and Aspects on Treatment
Fatigue is one of the most important long-lasting symptoms following TBI, and is most severe
immediately after head injury. However it is difficult to arrive at any clear figure as to how
common fatigue or, in particular, mental fatigue is. The reason for this is that different results
have been obtained, and these are attributable to differences in definitions and differences in
the methodology in the various studies. In follow-up studies, the frequency of prolonged
fatigue varies from 16 up to 73 % [4-6]. There is no correlation between persistent fatigue and
severity of the primary injury, age of the person at injury or time since injury [7, 8]. For those
suffering from fatigue 3 months after the accident the fatigue remained relatively stable during
longer periods [9]. In particular, for those subjects who were suffering from the syndrome one
year after the accident improvement in the fatigue was limited [10]
Mental fatigue is not an illness, rather it represents a mental sequel, probably due to a
disturbance of higher brain functions, either physical or psychological in origin. It is included
in, and defined within the diagnoses Mild cognitive impairment (F06.7), Neurasthenia (F48.0)
and Posttraumatic brain syndrome (F07.2) [11].
1.3. Typical characteristics of mental fatigue
A typical characteristic of pathological mental fatigue after TBI is that the mental exhaustion
becomes pronounced during sensory stimulation or when cognitive tasks are performed
for extended periods without breaks. There is a drain of mental energy upon mental
activity in situations in which there is an invasion of the senses with an overload of
impressions, and in noisy and hectic environments. The person feels that their brain is
overloaded after a tiny load. Another typical feature is a disproportionally long recovery
time needed to restore the mental energy levels after being mentally exhausted. The mental
fatigue is also dependent on the total activity level as well as the nature of the demands
of daily activities. Fatigue often fluctuates during the day depending on the activities carried
out. Thus, this fatigue is a dynamic process with variations in the mental energy level. The
fatigue can appear very rapidly and, when it does, it is not possible for the affected person
to continue the ongoing activity. Common associated symptoms include: impaired memory
and concentration capacity, slowness of thinking, irritability, tearfulness, sound and light
sensitivity, sensitivity to stress, sleep problems, lack of initiative and headache [12].
For many persons, this mental fatigue is the dominating factor which limits the person’s ability
to lead a normal life with work and social activities. For most people, fatigue subsides after a
period of time while, for others, this pathological fatigue persists for several months or years
even after the brain injury has healed. Interestingly, however is that as many as 30% of family
or friends interpreted fatigue as laziness [9].
Theories as to the mechanisms accounting for mental fatigue including our own theory,
suggest that cognitive activities require more resources and are more energy-demanding after
brain injury than usual [13, 14]. Thus, more extensive neural circuits are used in TBI victims
compared to controls during a given mental activity [15]. This indicates an increased cerebral
effort after brain injury.
Therapist Luann Jacobs describes mild TBI and the lack of energy and lack of endurance that
many can experience. As they are able to do what is normal and what appears normal, they
run the risk that their symptoms will be misunderstood [16].
“Mild brain injury is a real misnomer, as it conveys the idea that nothing much is a problem when quite the opposite is more often true. It is called “mild” because, in fact, the mildly brain injured can walk, talk, eat and dress independently, often times drive a car, shop, cook, go to school, or even work.
What the term fails to account for is the inherent limits of how often, for how long (endurance), and the all-important, how consistently (e.g., every day, once a week) these activities can be performed. Even more elusive is the concept of how many of these daily activities can be done sequentially in a given day as is normal in the lives of people who are not brain injured.
The fatigue they feel defies description, going far beyond and far deeper than anything a non-brain-injured person would consider profound exhaustion.”
From: A systematic review of fatigue in patients with traumatic brain injury: The course, predictors and consequences - ScienceDirect
Factors associated with fatigue
When we sought evidence of a temporal relationship between clinically important factors and fatigue we focused on: (1) TBI population characteristics (e.g. time since injury, severity of injury, comorbid conditions, etc.) and (2) our outcome of interest (i.e. fatigue) – its frequency, severity, and definition, with the goal of obtaining a set of risk factors that can be used for prognosis. Table 4 presents a descriptive summary of the available evidence. In summary, several potential risk factors for fatigue in TBI have been investigated, including those related to demographics and socioeconomic status, injury severity, medical comorbidities, baseline fatigue levels, genetic makeup, and physical and cognitive independence.
Fatigue at baseline, occurring at any time from injury through the acute care course, was found to be a primary predictor of symptom chronicity in TBI of varying severities (Norrie et al., 2010). Baseline fatigue was found to be one of the most powerful predictors of fatigue at follow-up (De Leon et al., 2009, Norrie et al., 2010). Other studies on chronic fatigue syndrome show similar associations between long-lasting fatigue and fatigue at baseline (Cairns and Hotopf, 1997, Nisenbaum et al., 2003, Kato et al., 2006), concurrent with the results of another study, where pre-stroke fatigue was reported to be related to fatigue in the acute phase after stroke (Lerdal et al., 2011). Despite reports of an impact of baseline fatigue on outcome at follow-up, the clinically important “critical values” of fatigue severity and duration following brain injury are not established. Future research should record frequent and specific data in investigation of the etiology of pre-morbid and baseline fatigue, and control for multiple factor interactions, in addition to the magnitude of effect attributable to individual factors in analyses. More attention needs to be paid to patients with intensive fatigue at baseline, as it is related to prognosis.
Fatigue is present before and immediately following injury, and can persist long term. The variation in findings supports the idea of fatigue in TBI as a nonhomogeneous entity, with different factors influencing the course of new onset or chronic fatigue. To decrease the heterogeneity, we emphasize the need for agreement on a core set of relevant fatigue predictors, definitions and outcome criteria.
Lundin et al. (2006) found poor memory, sleep disturbance and fatigue to be most commonly reported within their sample, with early symptom overlap correlated with later results. Similarly, Meares et al. (2011) found symptom overlap of fatigue, insomnia, and irritability at five days and three months post-injury, with some participants recovering from and others developing the symptoms as time went on.
Norrie et al. found a significant increase in the percentage of those with fatigue reporting depression and/or anxiety, both symptoms over the cut off indicating mild severity, at six months after injury, compared with reports at three months. This increase coincides with a leveling off of fatigue percentages. As fatigue becomes persistent, psychological factors such as anxiety and depression tend to worsen (Norrie et al., 2010).
Ponsford et al.’s mTBI group reported significantly poorer general health, vitality, and mental health, as demonstrated by their scores in the corresponding subscales of the SF-36, compared to trauma controls; however, a similar pattern was observed when participants completed the same scales but with regard to their pre-injury status. The authors highlighted the importance of documenting pre-injury status in TBI studies.
3.12.2. Studies with baseline assessment after one month post-injury
Bushnik et al., investigating changes in fatigue from 6 to 12 months post-injury, reported a significant change in the Pittsburgh sleep quality index (PSQI) scores: where fatigue increased, PSQI scores were higher compared to cases where there was no change or decreased fatigue. There were no other significant group differences on the pain VAS, disability rating scale, neurobehavioral functioning inventory motor subscale, the Craig handicap assessment and reporting technique (CHART) cognitive independence, and CHART occupation (Bushnik et al., 2008a). The authors suggested that, for TBI individuals who complain of fatigue, assessing sleep quality would be a high-yield correlate and possibly treatable with behavioral and/or medication interventions.
Kempf et al. reported no associations between fatigue parameters and TBI severity, alcohol intake at time of injury, nor with sleep duration, education, age or gender. They did, however, find a moderate correlation between FSS and depression symptoms assessed with the Beck depression inventory (BDI) (r = 0.46, p = 0.001), and with anxiety symptoms assessed with the Hospital anxiety and depression scale (HADS) (r = 0.37, p = 0.007). They also reported coincidence of fatigue and excessive daytime sleepiness (Kempf et al., 2010).
The APOE ɛ4 allele in persons with TBI was previously reported to be linked to an increased risk of Alzheimer's disease (Jellinger et al., 2001). In a study of the general population (O’Hara et al., 2005), a relationship between sleep apnea (SA) and dementia through the APOE ɛ4 allele was observed. Sleep apnea, highly prevalent in the TBI population (Mollayeva et al., 2013b), may explain the link between the APOE ɛ4 allele and fatigue and dementia. While studies to date have outlined the separate relationships between TBI, SA, fatigue, the APOE ɛ4 allele, and dementia, their complex interaction requires rigorous study.
Interpretation of scale items by the respondent can be significantly confounded by the association of fatigue with other symptoms, particularly apathy, excessive sleepiness, depression, lack of motivation, anxiety, litigation and cognitive dysfunction. In TBI patients, fatigue was reported to be associated with depression; moreover, in the regression analyses in one reviewed study (Norrie et al., 2010), the severity of fatigue was predicted by depression. Kempf et al. (2010) reported that fatigue and excessive daytime sleepiness coincided in their sample. Excessive daytime sleepiness may be an indicator of central nervous system (CNS) pathology due to brain injury (i.e. hypocretin/orexin deficiency), as well as related to quantity and quality of sleep (Mollayeva et al., 2013b, Baumann, 2012, Nardone et al., 2011, Mathias and Alvaro, 2012, Siebern and Guilleminault, 2012). While Kempf et al. (2010) studied and did not uncover a relationship between fatigue and sleep duration, quality of sleep was not investigated. A number of sleep disorders (i.e. sleep-related breathing disorder, periodic leg movement disorder, etc.) highly prevalent post-TBI (Nardone et al., 2011, Mollayeva et al., 2013c) are characterized by frequent arousals, which generally result in fragmented sleep, which can produce daytime sleepiness (Stepanski, 2002). Bushnik et al. (2008a) suggests assessment of sleep quality as a valuable measure when studying TBI patients with fatigue complaints.
Symptoms of fatigue and cognitive dysfunction have been reported to overlap in persons with TBI (Johansson et al., 2009, Zaben et al., 2013). This can potentially influence accuracy of self-report, as a person with cognitive dysfunction may not be able to fully grasp the changes in fatigue since their injury, as well as its impact on daily functioning, as required in completion of certain self-report measures. Bushnik et al. (2008a) reported that a subset of individuals who experienced significant increase in fatigue over the first two years post-injury demonstrated poorer outcomes in cognition, motor symptoms, and general functioning compared to those with decreased or stable fatigue (Bushnik et al., 2008a). A separate study had similar findings – subjective mental fatigue following brain injury was correlated with objectively measured information processing speed (Johansson et al., 2009). In other literature again, post-traumatic conditions such as hypopituitarism have been reported to have a wide range of manifestations, including fatigue, myopathy, cognitive difficulties, depression, and behavioral changes (Zaben et al., 2013). The same degree of fatigue, therefore, will not be perceived with equal intensity by persons with different comorbid conditions or fatigue etiology. Moreover, fatigue manifestation is thought to be differentially modulated by a variety of factors within and between TBI persons with time. Distinguishing fatigue as a result of TBI from fatigue associated with comorbid conditions (i.e. depression, pain, anxiety, apathy, sleep dysfunction, medication effect, etc.) is a complicated task. As such, future research should consider use of additional measures for common comorbidities when assessing PTF.
Fatigue severity (i.e. mean FSS scores) was higher in persons with TBI than previously reported for healthy adults (2.3 ± 0.7) (Krupp et al., 1989), but lower than those in patients with systemic lupus erythematosus (SLE) (4.7 ± 1.5) (LaChapelle and Finlayson, 1998), rheumatoid arthritis (4.2 ± 1.2) (Krupp et al., 1989) and psoriatic arthritis (6.9 ± 2.4) (Cella et al., 2005). Bushnik et al. (2008a) reported FSS scores obtained at 6, 12, and 18–24 months post-injury, all falling within the score range for non-fatigued control subjects (Table 5).
Studies differed in reports of fatigue severity over time, with some noting changes with and others stability. It is plausible that time since injury is a determinant of effectiveness of coping strategies and thereby perception of symptom severity. A study of persons with chronic fatigue syndrome (Brown et al., 2010) reported better adaptive coping strategies with longer disease duration.
CNS depressants can cause or increase fatigue (Liska, 2008). alcohol is a CNS depressant, and the injured brain is particularly sensitive to its effects at the highest centers (i.e. speech, thought, cognition) and lower brain functions (i.e. spinal cord reflexes, respiration), as the dosage increases (Liska, 2008). Norrie et al. (2010)
Intake of SSRIs was reported by 61.1% of the participants in Driver and Ede's study. While this class of medications is a first line of treatment for depression following TBI (World Health Organization, 2002), some drugs within this class (i.e. fluoxetine and paroxetine) may be problematic due to their adverse effects, including those related to fatigue and cognitive function (Schmitt et al., 2001). Another reviewed study reported use of opioids/opiates by 59.7% of participants (Meares et al., 2011). Opium alkaloids are narcotic analgesics and narcosis is defined by depression of the CNS leading to analgesia, drowsiness, changes in mood, mental clouding, lethargy, apathy and subsequent unconsciousness (Shukka and Devi, 2011). While currently there is no strong evidence directly relating physical and mental fatigue in TBI to side effects of opiates and opiods (Chapman, 2002, Leong and Royal, 2004), data on its safety for chronic use is also lacking (Rhodes, 2012). While our discussion of medication effects and fatigue in TBI is limited, given the complexity of the fatigue symptom and incomplete data available, future research should consider such effects, as the potential of medications to cross the blood–brain barrier and mimic neurological deficits and cause or exacerbate PTF, is real (Daneman, 2012, Maher et al., 2011).
None of the studies reviewed applied technologies (i.e. electroencephalography, functional magnetic resonance imaging, magnetic resonance spectroscopy, regional brain volumes, motor evoked potential, etc.) or markers of physiological processes (i.e. function of hypothalamic–pituitary–adrenal axis, autonomic nervous system response, metabolic processes, immune system response, etc.) to study the fatigue experienced by individuals with TBI. The latter is important, as research has shown that the resting pulmonary and cardiorespiratory function in patients with TBI is compromised (Jankowski and Sullivan, 1990). This can be related to deconditioning as a result of a more sedentary lifestyle (Giordon et al., 1998). In a study of maximal physiologic responses during exercise in patients with moderate to severe TBI at 17.2 ± 17 months after injury, several weeks of an exercise training program reduced physiologic fatigue (Bhambhani et al., 2005). Others reported that aerobic fitness in individuals with TBI enhanced cognition and improved mood (Carroll et al., 2004b, Cassidy et al., 2014). This is extremely important as fatigue perception ratings were found to be higher in patients with depression (Norrie et al., 2010). Again another study (Jankowski and Sullivan, 1990) reported that a 16-week circuit training program of moderate intensity and prolonged duration increased TBI patients’ oxidative capacity and muscular endurance and the index of physiologic fatigability was shown to be useful for the assessment and evaluation of individuals with TBI. Similarly, in a reviewed study by Driver and Ede (2009) fatigue elimination was reported after an eight-week group aquatic program, with no changes in fatigue in the control group. Thus, further study that accounts for the physiologic, objective performance, and/or homeostatic changes with regard to increased perception/manifestation of fatigue after brain injury is within the top priorities for future research.
fatigue as a symptom is nonspecific to TBI. Fatigue appears with other diagnostic labels in other clinical specialities – for example fibromyalgia, chronic fatigue syndrome, endocrine disorders, and patients with psychiatric illness (Norrie et al., 2010, Cairns and Hotopf, 1997, Nisenbaum et al., 2003, Kato et al., 2006, Lerdal et al., 2011, Cassidy et al., 2014).
Fatigue is a common symptom post TBI. Its frequency may change over time, but fatigue can persist years after the injury. This may be related to pre-morbid/early fatigue, mental health issues, other medical conditions, and ongoing societal stressors. Clinicians seeing patients with TBI at the acute stages post-injury with high early fatigue intensity, mental health issues, and litigation involvement should be aware that these may be associated with the development of persistent post-concussive symptoms. The available evidence on the associative value of these factors, as well as the consequences of fatigue, is currently not very strong, as we found just three cohort studies addressing these issues. More research is needed to establish associations between fatigue and other clinically important pre- and post-morbid variables (i.e. sleep dysfunction, depression, physical and cognitive impairments, other medical/neurological disorders), and their impact on outcomes post-injury. Medication effects, personal factors such as coping ability, physical deconditioning, stress level, and time factors should also be investigated. This is particularly important for translation of research into clinical practice, in order to address risk factors and course of condition. An international consensus, similar to the National Institutes of Health and developed for rehabilitation in TBI in 1999 (Consensus Conference, 1999) advising on how best to study clinically important symptoms such as fatigue in TBI, is of utmost importance. In particular, there needs to be a consensus on the definition of pathological PTF, set times for baseline assessment, recognizing the challenges in studying the symptom in moderate-severe brain injury at time zero, clinically relevant period of follow-up, acceptable attrition rates to ensure representative samples, and validated measures of outcome, all of which can reduce heterogeneity of results. What will be left to focus on then is the variety of lesions from TBI (i.e. white or gray matter, specific tract damage, lesion volume, localization of injury, etc.) and inter-individual variability in perception and multifactorial fatigue etiology, which may find study of individual patients best. A caveat to this point is that case-reports of patients whose symptoms and clinical course do not fit the typical picture, may lead to scientific progress in the understanding of and appreciation for the complexity of the fatigue symptom post TBI (Yennurajalingam and Bruera, 2007).
From: The neural correlates of cognitive fatigue in traumatic brain injury using functional MRI. - PubMed - NCBI
The present study used fMRI (functional magnetic resonance imaging) to objectively assess cognitive fatigue in persons with traumatic brain injury (TBI). It was hypothesized that while performing a cognitive task, TBI participants would show increased brain activity over time, indicative of increased cerebral ‘effort’ which might manifest as the subjective feeling of cognitive fatigue.
Methods and procedures: Functional MRI was used to track brain activity across time while 11 TBI patients with moderate–severe injury and 11 age-matched healthy controls (HCs) performed a modified Symbol Digit Modalities Task (mSDMT). Cognitive fatigue was operationally defined as a relative increase in cerebral activation across time compared to that seen in HCs. ROIs were derived from the Chauduri and Behan model of cognitive fatigue.
Main outcomes and results: While performing the mSDMT, participants with a TBI showed increased activity, while HCs subsequently showed decreased activity in several regions including the middle frontal gyrus, superior parietal cortex, basal ganglia and anterior cingulate.
Conclusions: Increased brain activity exhibited by participants with a TBI might represent increased cerebral effort which may be manifested as cognitive fatigue. Functional MRI appears to be a potentially useful tool for understanding the neural mechanisms associated with cognitive fatigue in TBI.
https://www.sciencedirect.com/science/article/pii/S0149763414002772bib0495
3ib) Fatigue part 2
Fatigue (as Disease), Interaction with
SubstancesFatigue from TBI and Factors
contributing to chronic fatigue
after traumatic brain injuryGene Interaction with Fatigue
(as Disease)Traumatic Brain Injury, Panhypopituitarism and Hormonal Evaluations as a Standard of Care
http://www.nevadaosteopathic.org/attachments/article/33/Clearfield%20Panhypopituitarism%20and%20TBI.pdf
Constant fatigue is the #1 symptom across TBI
100% of TBI patients experience fatigue. From there, 68% reported fatigue at 2 years post-injury, at 5 years post-injury 73% reported problems with fatigue.
From: The neural correlates of cognitive fatigue in traumatic brain injury using functional MRI. - PubMed - NCBI https://www.ncbi.nlm.nih.gov/pubmed/19408165)
The present study used fMRI (functional magnetic resonance imaging) to objectively assess cognitive fatigue in persons with traumatic brain injury (TBI). It was hypothesized that while performing a cognitive task, TBI participants would show increased brain activity over time, indicative of increased cerebral ‘effort’ which might manifest as the subjective feeling of cognitive fatigue.
Methods and procedures: Functional MRI was used to track brain activity across time while 11 TBI patients with moderate–severe injury and 11 age-matched healthy controls hCs) performed a modified Symbol Digit Modalities Task (mSDMT). Cognitive fatigue was operationally defined as a relative increase in cerebral activation across time compared to that seen in HCs. ROIs were derived from the Chauduri and Behan model of cognitive fatigue.
Main outcomes and results: While performing the mSDMT, participants with a TBI showed increased activity, while HCs subsequently showed decreased activity in several regions including the middle frontal gyrus, superior parietal cortex, basal ganglia and anterior cingulate.
Conclusions: Increased brain activity exhibited by participants with a TBI might represent increased cerebral effort which may be manifested as cognitive fatigue. Functional MRI appears to be a potentially useful tool for understanding the neural mechanisms associated with cognitive fatigue in TBI.
From: Clearfield Panhypopituitarism and TBI
http://www.nevadaosteopathic.org/attachments/article/33/Clearfield%20Panhypopituitarism%20and%20TBI.pdf
80% of TBI Injuries are mild without LOC
Acute hormone deficiencies occur in 56% of Head Injuries
36% continue on to Chronic Hormone Deficiency
Psychotropic Meds Mask Symptoms
– Psychotropic meds do not address underlying cause
Plan: Replace Deficient Hormones to Physiologic Levels
Psychotropic Meds Mask Symptoms
– Psychotropic meds do not address underlying cause
Plan: Replace Deficient Hormones to Physiologic Levels
From: https://www.ncbi.nlm.nih.gov/pubmed/23547621https://www.ncbi.nlm.nih.gov/pubmed/23547621
Trauma is responsible for sudden biochemical changes occurring at the time of impact, and the severity of brain insult can be graded by measuring these biochemical modifications, specifically, ROSmediated damage, energy metabolism depression, alteration of gene expression, and ultimately variation of NAA concentration, a surrogate marker of neuron dysfunction. TBI combines mechanical stress to brain tissue with an imbalance between CBF and metabolism, excitotoxicity, edema formation, and inflammatory and apoptotic processes. Understanding the multidimensional cascade of injury offers therapeutic options including the management of mechanical (hyper) ventilation, kinetic therapy to improve oxygenation and to reduce intracranial pressure, and pharmacological intervention to reduce excitotoxicity and intracranial pressure. The unpredictability of the individual’s pathophysiology requires monitoring of the injured brain to tailor the treatment according to the specific status of the patients. It will be important to better facilitate bidirectional translational research between preclinical and clinical investigators, which should serve to improve both approaches to animal modeling and the design of clinical trials. Future advances in clinical data sharing should improve TBI classification in ways that may lead to delineation of specific patient subgroups that may benefit from better targeted neuroprotective strategies.
From: http://www.psychiatrictimes.com/neuropsychiatry/neuropsychiatriceffectstraumaticbraininjury
Contusions are areas of cerebral bruising particularly involving gray matter, whereby blood leaks into the extravascular space. The contusion results in cell death and local loss of tissue. Diffuse axonal injury affects white matter anywhere throughout the cerebrum and brain stem. It may be followed by generalized atrophy with ventricular enlargement (Figure 2); this may take a few weeks or months to develop. Diffuse axonal injury in the brain stem is usually responsible for the slurred speech and severe ataxia that are seen in some severely disabled patients after TBI. Contusions and diffuse axonal injury may be complicated by anoxic brain injury that may occur soon after trauma because of poor cerebral perfusion secondary to raised intracranial pressure and focal strokes. In some patients, localized infarction occurs (Figure 3).
From: Oxford Textbook of Neurorehabilitation
edited by Volker Dietz, Nick Ward
CHAPTER
The applicability of motor learning to neurorehabilitation
John W. Krakauer
Note: citations excluded, as references unavailable without purchase (transcribed from google books preview)
A combination of biological psychological and social factors contribute to fatigue in all patients though the relative contribution of each varies between diagnosis and patient, failure to consider each sphere can lead to suboptimal treatment in all. Obvious examples are failure to consider depression in an individual who has prominent fatigue following a stroke, or overlooking medication side effects in a patient with CFS. Psychosocial issues, including unhelpful health beliefs, are particularly important to identify in neurological disease as these may be the most modifiable maintaining factors. Though divisions are fluid, in this section potential contributions to fatigue will be separated into primary and secondary factors. The former are directly attributable to the neurological disease process, while the ladder are physiological, psychological or behavioral changes occurring as direct or indirect consequences. CFS is an example of a condition in which extreme fatigue exist in the absence of overt neurological pathology, but the presence of immunological and endocrine abnormalities is well established. this demonstrates both the arbitrary separation between primary and secondary factors and the potentially profound impact that the latter can have. Effective treatment for CFS suggests what may improve fatigue in other conditions.
Neurological disorders are due to abnormalities of the structure or function of the nervous system. Those associated with fatigue affect diverse brain regions, however, and fatigue is also prominent in medical and psychiatric conditions, in which brain structural abnormalities are subtle or absent. Consequently fatigue is unlikely to be localized to a discrete brain region.
Early structural magnetic resonance imaging (MRI) studies of MS did not find any correlation between subjective fatigue and lesion load or brain atrophy, although recently associations between fatigue and volume loss have been reported, with atrophy in the striatum, thalamus, frontal cortex, and parietal cortex particularly highlighted. In a crosssectional study poststroke fatigue was more common in stroke then transient ischaemic attack (TIA) patients, suggesting at least some poststroke fatigue might be attributable to brain damage. Systematic review found no association between fatigue and white matter lesions or brain atrophy, however, although some studies did report an association with in infratentorial or basal ganglia stroke. No TBI studies examine correlations between structural abnormalities with fatigue, but clinical markers of injury severity do not predict fatigue. A study in patients who had had penetrating TBI found that fatigue was associated with ventromedal prefrontal cortex damage. Reduced gray matter volume is reported in CFS, with increased prefrontal cortex volume following treatment with cognitive behavioral therapy (CBT). In summary, when structural abnormalities are identified they implicated involvement of frontal and subcortical brain regions in fatigue.
As fatigue likely involve distributed brain regions, functional imaging may provide greater insights into its mechanisms. These approaches generally support the concept that fatigue is associated with dysfunction of corticalsubcortical circuitry, particularly circuits involved in attention and executive function. in MS there is decreased regional glucose metabolism in the frontal cortex and basal ganglia of fatigue patients. In TBI brain activity is increased in the middle frontal lobe, basal ganglia, and anterior cingulate during a speeded cognitive task; in PD decreases in frontal lobe perfusion are greater in patients with fatigue than those without (which was associated with executive function impairments); and CFS patients had differing patterns of activation of prefrontal cortical regions compared to healthy controls. Functional Imaging studies of fatigue stroke patients have not been undertaken, but poststroke fatigue has been related to attentional and executive impairment.
In summary, convergent data across neurological conditions suggest dysfunction in the striatalthalamicfrontal system is important in fatigue. These impairments may necessitate higher levels of mental effort for complex tasks, which increases subjective fatigue. In conditions with damaged brain structure (e.g. MS, TBI) recruitment of expanded pools of cortical neurons likely reflects brain plasticity unmasking latent pathways. Though adaptive, it maybe energy intensive, excessive use of neuronal pools results in fatigue. In CFS disruption again seems present but likely arises through different routes, which may include mechanisms such as sustained abnormalities of attentional focus
Inflammation is associated with fatigue, as evident from the lethargy of acute infections. This is mediated by proinflammatory cytokines, which act on the brain to result in drowsiness, loss of appetite, decreased activity and withdrawal from social interaction. The association between treatment with interferona (IFNa) and fatigue (which is dissociable from depression) is well recognized. As inflammatory degenerative disorders, elevated cytokines are particularly relevant to fatigue in MS and systemic lupus erythematosus (SLE). Cytokines are however also elevated poststroke and TBI, in CFS, and even in PD, and depression, likely also contributing to fatigue in these conditions.
Alterations in the hypothalamicpituitaryadrenal (HPA) axis are among the most replicated findings in CFS, mild hypocortisolaemia being consistently reported and attributed to enhanced negative feedback in the HPA axis. This contrasts with the increased HPA axis activity and raised cortisol levels seen in depression. Whereas hormonal changes are relatively subtle in MS and CFS, in TBI, (and obviously pituitary stroke), they can be gross and necessitate replacement treatment. In TBI these abnormalities are not restricted to the acute phase, with as many as 25% of longterm survivors showing one or more pituitary hormone deficiencies. As well as hypocortisolaemia and hypothyroidism being obvious causes of fatigue, an association with lowered growth and sex hormone levels following TBI has been reported.
The possibility of additional medical pathology must be remembered. There should be blood screens for common hematologic and metabolic conditions and thyroid dysfunction. Recommended investigations to a diagnosis of CFS, exclude other conditions and ‘red flags’ for alternative diagnostic explanations are shown in Table 26.3. in MS vitamin D deficiency is common. Though and association with fatigue was not found in MS it had been in the general population; consequently, assessment of 25hydroxy vitamin D levels may be considered. Infections can worsen fatigue and should be excluded. An MS exacerbation may present as fatigue prior to clinical manifestation.
Medications frequently causing fatigue include antispasticity agents (EG baclofen or tizanidine), narcotic analgesics, sedative hypnotic or anticonvulsant agents, sedative antidepressants or anxiolytics, and antihypertensive medications. Patients often report increased fatigue with IFN therapy, though fatigue often improves with time on interferon. Pretreating with non steroidal antiinflammatory may improve IFN associated fatigue. Hypertension or hypotension secondary to excessive antihypertensive use may be associated with post stroke fatigue, though whether there is causal relationship is uncertain.
From: https://www.healthyplace.com/blogs/anxietyschmanxiety/2016/03/tbicananxietydisorderscomefromabraininjuryhttps://www.healthyplace.com/blogs/anxietyschmanxiety/2016/03/tbicananxietydisorderscomefromabraininjury
From: Can Anxiety Disorders Come from a Traumatic Brain Injury| HealthyPlace
Traumatic brain injury (TBI) is the major cause of death and disability among young adults. In spite of preventive measures, the incidence of a TBI associated with motor vehicle accidents, falls, assault, and highcontact sports continues to be alarmingly high and constitutes a major public health concern. In addition, the recent military operations in Iraq and Afghanistan have resulted in a large number of persons with blast injuries and brain trauma. Taking into account that cognitive and behavioral changes have a decisive influence in the recovery and community reintegration of patients with a TBI, there is a renewed interest in developing systematic studies of the frequency, mechanism, and treatment of the psychopathological alterations observed among these patients.
Nonmilitary TBI
Each year, an estimated 1.5 million Americans sustain a TBI that requires hospitalization. As a result of these injuries, 80,000 to 90,000 patients experience longterm disability.1 There is consensus that cognitive, emotional, and behavioral problems constitute the major source of disability for TBI patients.
The neuropsychiatric consequences of a TBI can be studied from a dimensional perspective using neuropsychological tests and behavioral scales that have been extensively validated in acutecare settings and rehabilitation services.25 Cognitive and behavioral morbidity can also be assessed from a categorical, diseasebased perspective, which assumes that psychiatric disorders, although diagnosed through a recognized constellation of symptoms, have an identifiable biological substrate, a distinct clinical prognosis, and an expected treatment response.6
Fann and colleagues7 examined the risk of psychiatric illness after a TBI in an adult health maintenance organization population. They compared the frequency of psychiatric disorders among 939 patients with a TBI and 2817 controls. The prevalence of any psychiatric illness in the first year was 49% following a moderate to severe TBI, 34% following a mild TBI, and 18% in the control group. Among study participants without a history of psychiatric illness, those with moderate to severe TBI were 4 times (95% confidence interval [CI], 2.4 to 6.8) more likely to sustain any psychiatric illness in the 6 months following a TBI than those without a TBI. The risk was 2.8fold higher (95% CI, 2.1 to 3.7) in patients with a mild TBI compared with study participants who did not have a TBI.7
Mood disorders are the most frequent psychiatric illness observed among patients with a TBI.810 Hibbard and colleagues11 used a structured interview and DSMIV criteria to identify Axis I psychopathology in 100 adults with a TBI who were evaluated, on average, 8 years after trauma. The prevalence of major depression in this population was 61%. More recently, Kreutzer and colleagues12studied the prevalence of major depressive disorder in a sample of 722 outpatients with a TBI, evaluated an average of 2.5 years following brain injury. Major depression, defined using DSMIV criteria, was diagnosed in 303 patients (42%). Findings from a prospective study indicate that the frequency of mood disorders was significantly greater in patients with a TBI than in a control group of patients who had had an orthopedic trauma. A mood disorder developed at some time during the first year after injury in 46 of 92 patients with a TBI (51%), compared with 6 of 27 patients (22%) with multiple traumatic injuries but without CNS involvement. In addition, the frequency of major depressive disorder was significantly greater in patients with TBI than in the control group.13 Thus, mood disorders were significantly more frequent in patients with a TBI than in patients with similar background characteristics who underwent similar levels of stress (eg, motor vehicle accidents) but who did not sustain brain injury. This suggests that structural brain damage associated with a TBI constitutes an important contributing factor to the development of affective disorders. Furthermore, patients who experience major depression following a TBI frequently show structural and/or functional alterations in the prefrontal cortex as evidenced by abnormal performance on neuropsychological tests or by abnormal neuroimaging findings.1315
Emotional processing and mood regulation involves the complex interaction between prefrontal regions (eg, anterior cingulate gyrus, orbitofrontal cortex) and limbic structures (eg, amygdala, hippocampus, ventral striatum). Different forms of traumatic lesions such as diffuse axonal injury and cerebral contusions may result in disruption of these neural circuits and, consequently, in affective disturbance. Furthermore, these changes may persist and evolve with time.
A significant proportion of patients in whom mood disorders develop following a TBI will progress to a more chronic and recurrent form of these psychiatric conditions, spanning many years. Thus, the prevalence of psychiatric disorders continues to be significantly higher in TBI patients than in control groups many years after the traumatic injury.1618
A recent community study suggests an association between a history of a TBI and an increased lifetime prevalence of major depression.17 The investigators found that the lifetime prevalence of major depression among men who had sustained a TBI during the Second World War was 18.5% versus 13.4% for a comparable group without a TBI. Koponen and colleagues18 assessed the frequency of Axis I and Axis II disorders in a group of 60 patients 30 years after sustaining a TBI. The patients showed a lifetime prevalence of major depression of 26.7%. Overall, these findings suggest that patients with a TBI have recurrent depressive disorder throughout their lifetime at a significantly higher frequency than comparable patients without a TBI. When present, affective disturbances have a large impact on family relationships, social integration, and return to productive activity.
Anxiety disorders occur in a significant proportion of patients with a TBI and frequently coexist with depressive disorders (Table 1).18,19 There is a significant degree of comorbidity between mood and anxiety disorders among patients with a TBI. For example, about twothirds of patients in whom major depression develops also meet diagnostic criteria for generalized anxiety disorder.13
Posttraumatic stress disorder (PTSD) is another frequent psychiatric complication in patients with traumatic injuries.20 Whether unconsciousness and posttraumatic amnesia associated with a TBI would preclude the onset of PTSD in patients who have had a lifethreatening experience, such as a motor vehicle accident or physical assault, has been widely debated. In fact, PTSD has been described in patients with a TBI of different degrees of severity, even among those patients who have partial or fragmentary recollection of the contingencies of the traumatic episode.2123 Recently, Glaesser and colleagues24 assessed the occurrence of PTSD in a group of 46 patients with a TBI who were admitted to an acute neurorehabilitation clinic. They concluded that a TBI and PTSD are not mutually exclusive. However, PTSD was unlikely to develop in victims of accidents if trauma had resulted in a prolonged period of unconsciousness. Bryant and colleagues25 analyzed the relationship between resting heart rates at 1 week and 1 month following a severe TBI and a PTSD diagnosis assessed 6 months after the injury. In these trials, PTSD developed in 16 of 68 (23%) severely injured patients. Compared with patients in whom PTSD did not develop, the patients had significantly higher heart rates at 1 week but not at 1 month after trauma. These findings suggest that fear conditioning can occur independently of the level of awareness and contribute to the onset of PTSD. These investigators also reported that patients who had PTSD at 6 months following a severe TBI had significantly poorer functional and vocational outcomes than did patients who did not have PTSD.26
From: https://www.healthyplace.com/blogs/anxietyschmanxiety/2016/03/tbicananxietydisorderscomefromabraininjury
Anxiety disorders, like all mental illnesses, are disorders of the brain. The brain is an organ of the body, and it can experience disease and/or injury just like any other organ. Traumatic brain injury can cause serious damage inside the brain. Depending on what part of the brain is damaged, TBI can lead to mental illness. Anxiety disorders can, indeed, come from a traumatic brain injury
Also, the very symptoms of brain injury can be so disruptive, so bothersome to someone’s life, that they cause anxiety. The symptoms of a traumatic brain injury include:
• Concentration difficulties
• Memory problems
• Organizational and/or planning issues
• Decisionmaking difficulties
• Being easily overstimulated by crowds, noises, etc.
• Increased susceptibility to pressure and stress
• Fear and worry that others will judge you as stupid, incompetent, unreliable
• Worry about making mistakes
• Anxiety about the brain injury symptoms themselves
Hope for Anxiety Disorders that Come From Traumatic Brain Injury
The human body possesses an amazing ability to heal. To be sure, many times brain injury involves permanent damage. However, a degree of healing does occur, and people have remarkable capacity to transcend their difficulties, to adjust, and to fix what can be fixed (Brain Change And PTSD: Proof Recovery Is Possible). Anxiety is very treatable (unfortunately, it’s not always quick and easy, but it is, indeed, treatable).
Some things that people can do when faced with anxiety disorders that come from traumatic brain injury include:
• Rest, rest, rest – that’s how the brainand with it, anxietyheals.
• Attend to your environment (reduce noise, keep light levels low).
• Practice selfcare.
• Establish a routine to reduce the need for decision making/concentrating and to lower stress.
• See medical doctors and follow their instructions, medications, etc.
• See a therapist to help deal with the anxiety.
My own experiences with anxiety came from a brain injury. I was anxietyprone before the TBI, but the actual anxiety disorders developed postTBI ditto bipolar 1 disorder. Here, too, I had mild symptoms before the injury, but the brain injury exacerbated them. I know firsthand that TBI can contribute to mental illness. That also means that I know that people can transcend their troubles, rising above even mental illness and brain injury. I am sincere when I write that there is hope for anxiety disorders that come from brain injury.
From: Phoenix rising user 'MishMash' http://forums.phoenixrising.me/index.php?threads/headinjuryandhypopituitarism.22717/
Head injuries can cause collapse or damage to the pituitary gland. Which can be seen in a scan (sometimes). This usually correlates, but not always, to symptoms of low pituitary function.
But the number of people who have collapsed pituitary gland, and hence low pituitary function, due to accidents, head injuries is tiny compared to the number who get it idiopathically. Collapsed, or disappearing, pituitary gland is known as "empty sella syndrome," because the gland sits naturally in a bone cavity called "the sella."
ESS is very common among CFS patients BTW. Before he left, Cort did an interesting feature on this for PR. A number of members posted they had ESS. Cort himself said the radiologist noted that his pituitary gland had disaappeared. One of the things he was recommending as a CFS spokesman was further research into this.
The chances for getting ESS go up markedly as age increases. Intracranial hypertension (too much brain fluid pressure) is usually cited as the reason for young people getting it. Too much brain fluid pressure has been associated with connective tissue weakness (or EhlersDanlos) since the cerebral ducts are weak and floppy, and get kinked and clogged easily. So the fluid just keeps building up in your cranium. Which leads to lower blood flow to the brain, if the theory is correct.
Some people go as far as putting in shunts. I've done quite a bit of research on this, and the shunts rarely result in reduction in symptoms, from what I've read. The doctor at "prettyill.com" has recommended taking diuretics to reduce cerebral brain fluid amounts; hence reducing pressure on the brains, and lowering frequency of headaches.
From: http://www.psychiatrictimes.com/neuropsychiatry/neuropsychiatriceffectstraumaticbraininjury
Neuropsychiatric assessment
The neuropsychiatric assessment starts by evaluating the severity of brain injury. In this way, the likely outcomes attributable to direct effects of brain injury can be determined, and any mismatch between these and what is observed can be attributed to psychological reactions or independent events. So, for example, in somebody with a severe psychotic illness that develops 3 months after an injury with no loss of consciousness, one can be fairly confident that the illness is not a direct consequence of the effects of brain injury on delusion formation. It is possible that the psychological trauma of the injury has allowed an acute psychotic reaction, or even that the injury was irrelevant and that the person was on the path to becoming schizophrenic anyway. On the other hand, it is likely that the psychotic illness is a direct effect of the brain injury in somebody in whom a delusional misidentification syndrome develops 3 months after an injury that was followed by coma for a week and delirium for several weeks.
The severity of brain injury is measured by the following:
Glasgow Coma Scale (used soon after injury)
Duration of loss of consciousness
Duration of posttraumatic amnesia (PTA), ie, the interval between the injury and the return of continuous daytoday memories
The duration of PTA is particularly useful as a measure of the severity of the brain injury because it can be measured retrospectively, eg, in the clinic years after injury, and it is a good predictor of outcome.3 As a rule, if PTA lasts less than 1 week, a reasonably good outcome is expected. If PTA lasts longer than 1 month, significant disability is likely; a good proportion of those affected will not be able to return to work or to independent living. In general, younger individuals (those in their late teens or 20s) tend to do much better.
An MRI scan is essential in cases where the extent of damage is unclear because it may show unexpected brain injury. Gradient echo sequences are the most sensitive and should be undertaken, particularly in those with mild injury. A normal MRI scan does not rule out brain injury, but it does make significant disability as a direct effect of severe brain damage unlikely. Electroencephalography is usually not helpful, even as a predictor of posttraumatic epilepsy.
Neuropsychometric assessment can be useful in defining the severity of cognitive impairment and any areas of particular impairment. Such tests as the North American Adult Reading test are available and provide an estimate of the patient’s preinjury IQ. Such assessment is necessary for the accurate interpretation of a patient’s postinjury performance. Also, make sure that tests of executive function have been done. Note, though, that normal neuropsychometric test results do not rule out brain injury as the cause of problems with executive functions in everyday life.
Cognitive and behavioral sequelae
Irritability and aggression are probably the most common behavioral consequences of TBI. However, it can be difficult to know the extent to which brain injury is a factor in aggressive behavior. Many patients who have sustained a TBI were prone to antisocial behavior before the injury. It is sometimes argued that because a behavior is sensitive to psychological cues, it is not the result of a brain injury; however, this is not correct. For example, just because extreme aggression is seen only in the context of the family and never at work does not necessarily mean that the aggressive behavior is unrelated to a brain injury.
Teasing out the role of brain injury can be difficult and usually relies on a good objective history of the behavior before and after the injury, along with an assessment of the likelihood of significant brain injury, which depends in part on the location of the injury. Cold, goaldirected aggression is seen in patients with psychopathic personality disorder, whereas the aggression of the braininjured patient is usually impulsive and quite out of proportion to the trigger. The latter, when severe, may be described as episodic dyscontrol syndrome. But in practice, it is not easy to discern the origins of aggression (constitutional vs brain injury) from its phenomenology.
In patients with severe brain injury, a typical clinical picture consists of significant cognitive impairment, particularly in the domains of attention and concentration, psychomotor speed, memory, and executive function, as well as fatigue and problems with motivation. The patient is likely to be selfcentered, thoughtless, and crude in social relationships. He or she may show disinhibited behavior that is often sexual. Agitation and repetitive purposeless behaviors may also be present. Lability of mood is common; patients are often described as childish or moody.
Mild traumatic brain injury
Chronic symptoms of a rather nonspecific nature are quite common after mild TBI (Glasgow Coma Scale score of more than 12; loss of consciousness, less than 30 minutes; PTA, less than 24 hours). Approximately onehalf, onequarter, and oneeighth of patients still have significant symptoms at 3, 6, and 12 months, respectively.4 Typical symptoms include headaches, fatigue, dizziness, depression, and difficulties with concentration and memory, which are often complicated by anxiety symptoms related to travel and posttraumatic stress disorder. Alteration of cerebral blood flow during working memory tasks 1 month after injury and longstanding changes in fractional anisotropy—an MRI measure of white matter integrity—have been shown.5
These postconcussional symptoms are nonspecific and are seen almost equally often in patients with musculoskeletal injuries but no head injury and in patients with chronic pain or chronic fatigue syndrome. In some patients with longstanding postconcussional symptoms, the extent and severity of the symptoms suggest that the illness is a form of somatization disorder. Perhaps the best model is that symptoms soon after injury are secondary to the direct effects of trauma to the head and brain, but that over time psychological factors intervene to prevent a healthy recovery.6
Management of patients with TBI
Agitation in the early postinjury period (eg, days or weeks after a severe injury) is common and usually selflimited. It is often associated with disinhibited behavior, particularly sexual disinhibition. There may be evidence of delirium and the patient is likely to show poor orientation and poor insight.
The development of agitation is a warning that something may not be right physically. For example, the patient may have thrown off some fat emboli from a fractured femur or be in urinary retention. Agitation may also be the first sign of infection entering through a cerebrospinal fluid leak. The first stage of management is a review of the patient’s medical and surgical recovery. Intoxication from medication may be to blame, and in some, agitation is a manifestation of craving because of substance abuse at the time of the injury.
Management rests on principles similar to those used in patients with delirium. Family and caregivers may be a helpful resource if they can spend time with the patient. The main task is to ensure the safety of the patient and others, monitor the patient’s physical recovery and prescriptions, and wait for improvement.
In the longer term (months and years after injury) aggression can be a major disability. It can cause severe family/caregiver burden and can interfere with a return to work. A good rehabilitation program can help by improving engagement in activities and selfconfidence. Specific anger management techniques should be tried, but they do not always work.
Drug treatment should not be started at the first sign of agitation and aggression. Wait before starting drug therapy and, if possible, get repeated baseline measures of severity to see whether the problem persists. Symptoms wax and wane, often for no identifiable reason, and improvements can be attributed to the medication when in fact they were merely the result of the passage of time.
It is not possible to provide good evidencebased guidance on which drug to choose to manage agitation and aggression. Antidepressants, mood stabilizers, antipsychotics, and bblockers may all have a role. Which to choose may be determined by comorbid symptoms (eg, depression, seizure disorder, or anxiety disorder). When treating agitation or aggression with medication, beware of making things worse by increasing confusion; adding akathisia to the problem list; disinhibiting the patient; or, by using rapidacting anxiolytics, unwittingly reinforcing the behavior. A good resource when deciding which drug to use is provided by the Neurobehavioral Guidelines Working Group.7
CHECKPOINTS
In somebody with a severe psychotic illness that develops 3 months after a traumatic brain injury with no loss of consciousness, one can be fairly confident that the illness is not a direct consequence of the effects of the brain injury on delusion formation.
If posttraumatic amnesia (PTA) lasts less than 1 week, a reasonably good outcome is expected; if PTA lasts longer than 1 month, significant disability is likely.
In patients with severe brain injury, a typical clinical picture consists of significant cognitive impairment—particularly in the domains of attention and concentration, psychomotor speed, memory, and executive function—as well as fatigue and problems with motivation.
Longterm functional deterioration
After a severe injury, most recovery occurs during the first 1 to 2 years. The patient then reaches a plateau and may be left with significant disability. Generally, the disability that remains at about 2 years postinjury is relatively fixed. However, some longterm followup studies have shown that a proportion of patients continue to change, for better or for worse, for many years after injury.8,9 Three clinical scenarios for deterioration of symptoms are possible.
The patient “gives up.” After initial gains in the months or years after a severe injury, the patient becomes noncompliant with therapy and withdraws socially; rehabilitation gains are lost. This is possibly a consequence of the patient’s greater awareness of his very disabled state. The optimism of the early phases of recovery begins to be replaced by the realization that the rate of improvement has slowed and full recovery is unlikely. In my experience, it is not easy to help such patients.
Psychosocial factors affect the patient’s recovery. Compared with patients who do well, patients who deteriorate—up to 10 years postinjury—tend to be more anxious and depressed. They have more problems with alcohol (both before and after the injury), have lower selfesteem, and are more likely to have been injured in an assault.10
Dementia may ensue. Patients who have sustained a TBI may be at increased risk for Alzheimer disease or other dementias, or for a lesser degree of decline in cognitive function.11,12
Secondary complications of TBI need to be considered in any patient with functional deterioration (Table 1). Which medication to choose to reverse the deterioration may be a matter of trial and error. Antidepressants are a reasonable firstline therapy, particularly if there is evidence of depressive symptoms. If fatigue is prominent, modafinil may help; methylphenidate may help with concentration13; cholinergic agents may be worth trying in those with deteriorating cognition.14
Depression
The diagnosis of depression after TBI is not straightforward. The brain injury may have direct effects on the control of facial expression so that the patient looks depressed, regardless of how he is feeling. Brain injury can have direct effects on pathways involved in appetite, sleep, pleasure, and reward, resulting in biological symptoms of depression even though the patient may not be depressed. Symptoms of distress and sadness can be understood as a reasonable reaction to a desperate predicament. Other changes in mood control may be seen—most commonly lability of mood, but also alexithymia.
The assessment depends on understanding the severity of the distress and the degree to which it is intractable and enduring. Feelings of worthlessness, hopelessness, or guilt suggest that clinical depression may be present. Suicidal ideation is not uncommon, and rates of suicide after TBI are increased 2 to 3fold.
Psychotic symptoms
Once the patient emerges from coma after a severe brain injury, there may be days or weeks of delirium. As with any delirium, hallucinations and delusions are common. When the patient emerges from the delirium, more discrete psychotic symptoms may become apparent. Confabulation, delusional disorientation, and delusional misidentification are characteristic of these early psychotic symptoms.
Confabulations are often banal. For example, a patient may describe visits from friends or family when in fact there have been none. However, confabulations may be more bizarre, (eg, a patient recalls a helicopter evacuation from the ward next door at the same time that a gun battle took place on the hospital roof). Delusional disorientation is common and might typically involve descriptions of the ward as being the patient’s place of work. Or the patient may believe that he is on a ship at sea.
Delusional disorientation may overlap with reduplicative paramnesia, so the patient may believe that he is in an annex of the hospital but in another part of the country. Other delusional misidentifications may be seen, particularly the Fregoli syndrome: the patient is convinced, for example, that the old lady in the bed opposite him is his aunt. Another delusion, also based on attributing familiarity when in fact there is none, is the patient’s belief that he has seen you, the treating doctor, before, whereas in fact you have never met.
Remember that confabulatory delusional states are part of a resolving organic mental state. Symptoms are likely to improve. Antipsychotic drugs may have little effect and, particularly if there are no concerns about patient safety because of delusions, are probably not needed.
Psychotic states that develop in the longer term may still be directly attributable to the brain injury. For example, depersonalization may result in nihilistic delusion, or poor memory may result in delusions of persecution. Morbid jealousy may be seen and is particularly dangerous in somebody who has suffered a brain injury, given the risk of violence and poor impulse control. Whether TBI can produce a schizophrenialike psychotic disorder is uncertain.15 A study from Sweden suggests that TBI results in a slight increase of risk for nonaffective psychotic illness, not schizophrenia.16
From: http://www.psychiatrictimes.com/neuropsychiatry/neuropsychiatriceffectstraumaticbraininjury
In patients with severe brain injury, a typical clinical picture consists of significant cognitive impairment, particularly in the domains of attention and concentration, psychomotor speed, memory, and executive function, as well as fatigue and problems with motivation. The patient is likely to be selfcentered, thoughtless, and crude in social relationships. He or she may show disinhibited behavior that is often sexual. Agitation and repetitive purposeless behaviors may also be present. Lability of mood is common; patients are often described as childish or moody.In patients with severe brain injury, a typical clinical picture consists of significant cognitive impairment, particularly in the domains of attention and concentration, psychomotor speed, memory, and executive function, as well as fatigue and problems with motivation. The patient is likely to be selfcentered, thoughtless, and crude in social relationships. He or she may show disinhibited behavior that is often sexual. Agitation and repetitive purposeless behaviors may also be present. Lability of mood is common; patients are often described as childish or moody.
From time to time every psychiatrist comes across patients whose problems are at least in part related to the neuropsychiatric consequences (behavioral, cognitive, and emotional) of traumatic brain injury (TBI). TBI affects approximately 2 of every 1000 persons per year. Those who are vulnerable to mental illness (eg, persons with alcohol abuse or antisocial personality disorder) are particularly at risk. Patients with TBI often have poor insight and may need hospitalization for their own safety. The neuropsychiatric and other sequelae are longterm; a head injury is for life.
A telling illustration from 1937 by Courville, a neuropathologist, nicely demonstrates why TBI is of interest to psychiatrists (see figure 1 in Fleminger 20091). The illustration is a composite of the location of contusions found in 50 patients who died of TBI. The sites of specific vulnerability to contusions are the medial orbital frontal lobe and the anterior temporal lobes (Figure 1). Areas where contusions rarely occur include the primary motor, somatosensory, and visual cortex. Therefore, areas of the brain concerned with social function and decision making are particularly vulnerable.2 It is unsurprising that neuropsychiatric sequelae outstrip neurophysical sequelae as the major cause of disability after TBI.
Ut eget felis
Sed velit congue viverra. Sed porta mattis luctus. Curabitur feugiat pharetra sem eu iaculis. Phasellus venenatis volutpat arcu id placerat. Aliquam fringilla ligula eu purus lacinia at volutpat nunc malesuada. Nunc a augue ac orci tempus commodo.
3ic) Fatigue part 3
Molecular Functions Associated
With FatiguePathways Associated With Fatigue
Cognitive Fatigue
Traumatic Brain Injury, Panhypopituitarism and Hormonal Evaluations as a Standard of Care
http://www.nevadaosteopathic.org/attachments/article/33/Clearfield%20Panhypopituitarism%20and%20TBI.pdf
Constant fatigue is the #1 symptom across TBI
100% of TBI patients experience fatigue. From there, 68% reported fatigue at 2 years post-injury, at 5 years post-injury 73% reported problems with fatigue.

Mauris sit amet
Tortor, eget ornare urna. Duis varius tellus eros. Donec odio arcu, rutrum ac rutrum eget, bibendum ac enim. Phasellus hendrerit iaculis purus. Aliquam sit amet molestie odio. Sed commodo dictum consequat aenean in est.
Ut eget felis
Sed velit congue viverra. Sed porta mattis luctus. Curabitur feugiat pharetra sem eu iaculis. Phasellus venenatis volutpat arcu id placerat. Aliquam fringilla ligula eu purus lacinia at volutpat nunc malesuada. Nunc a augue ac orci tempus commodo.
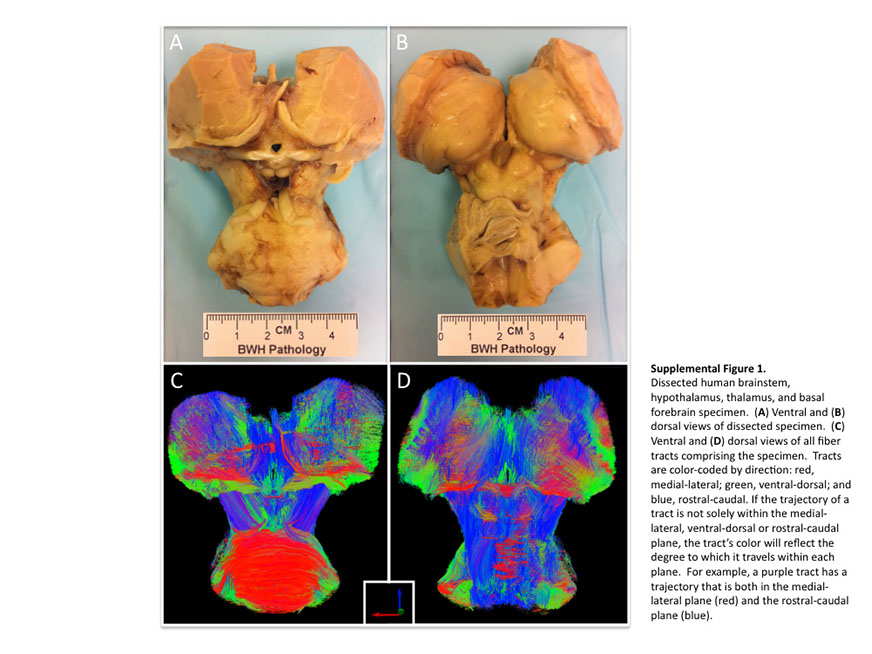
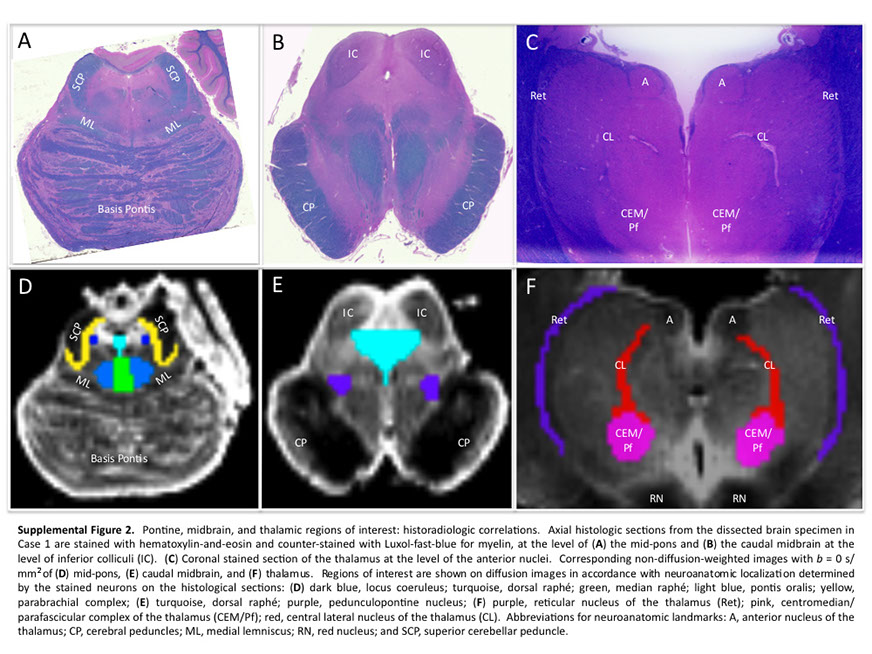
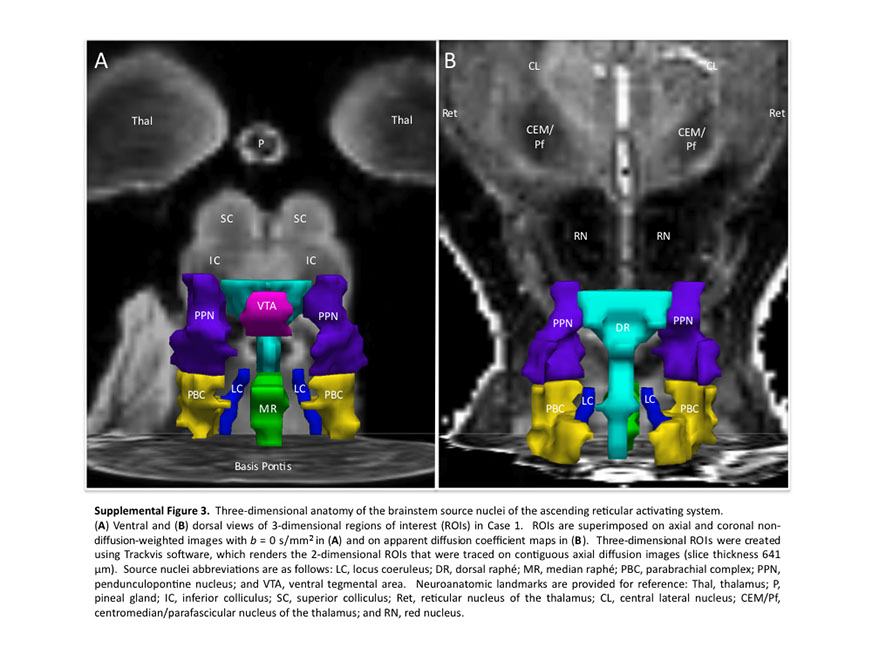
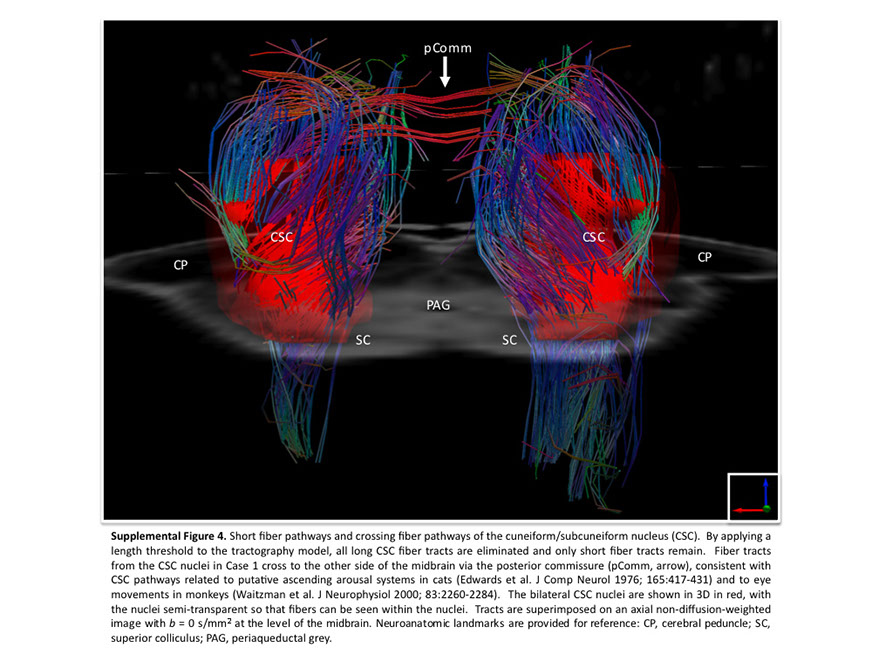
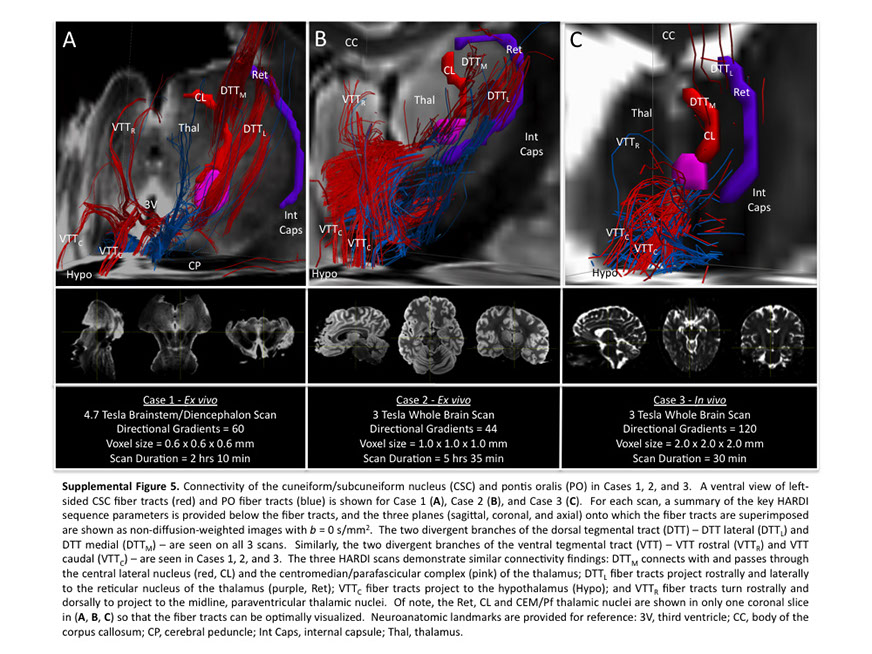
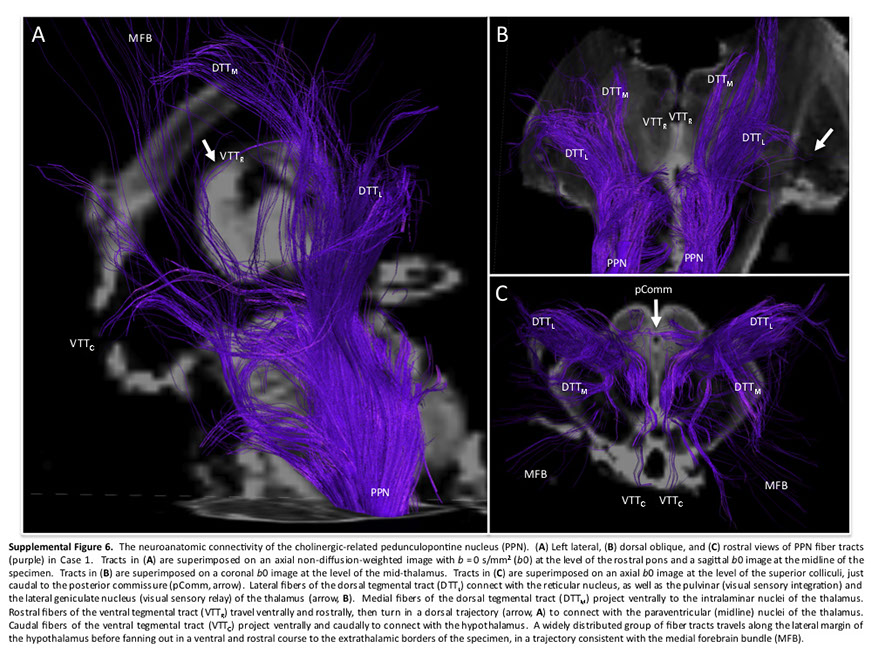
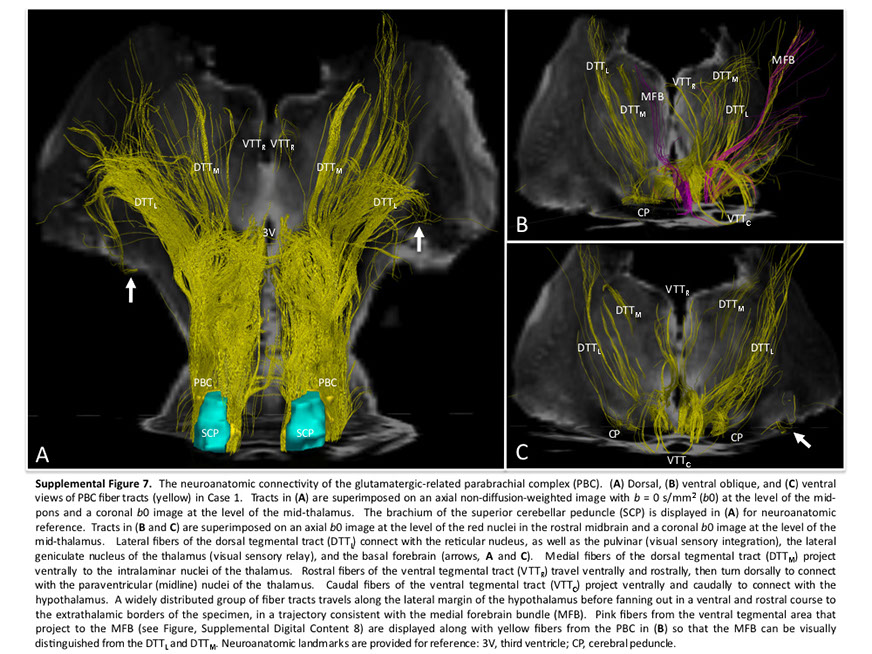
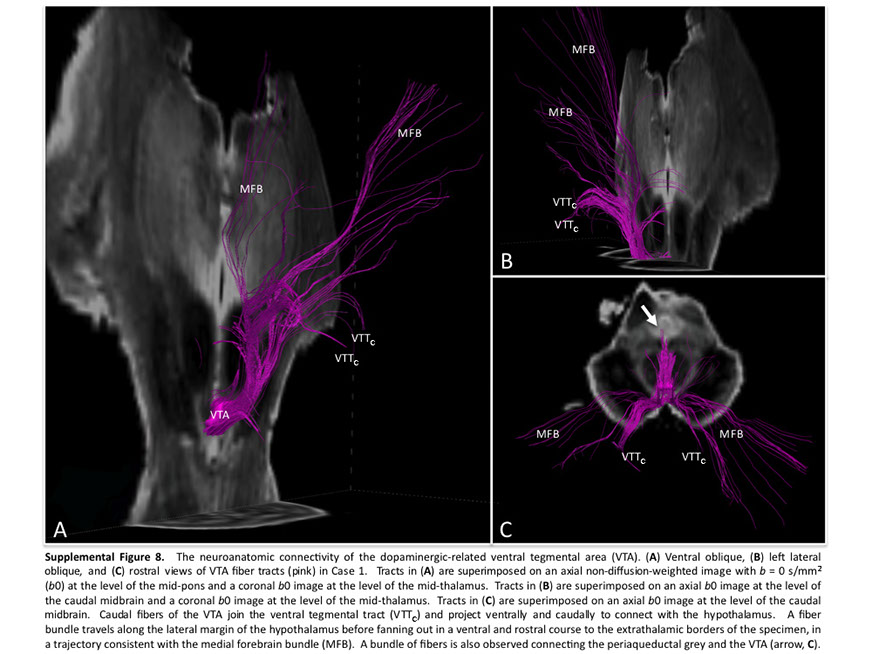
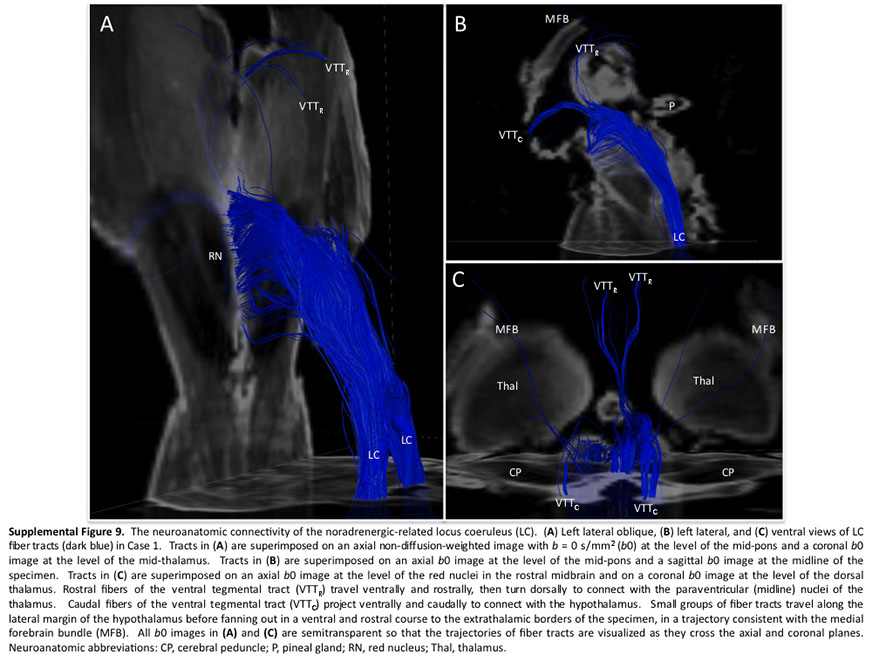
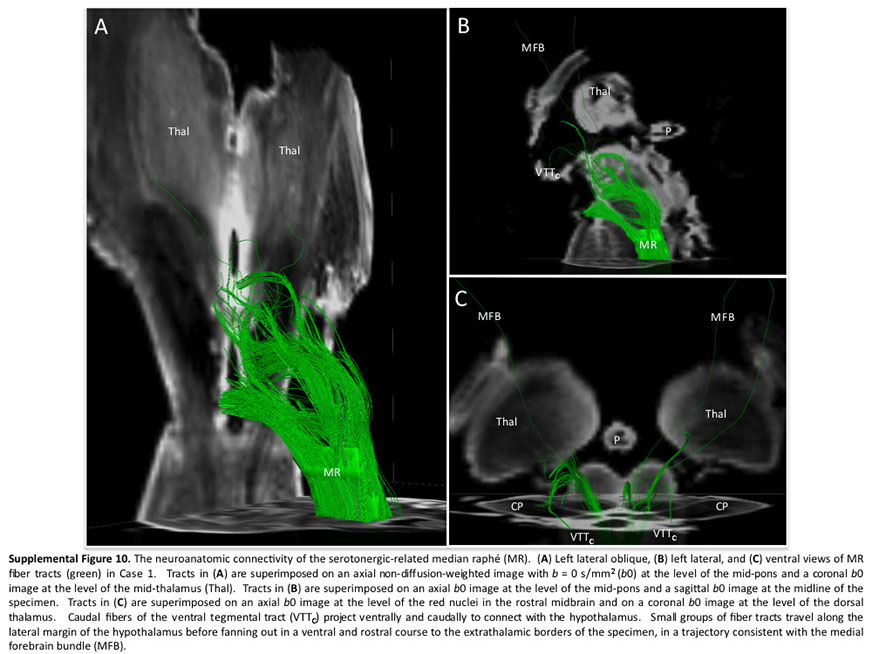
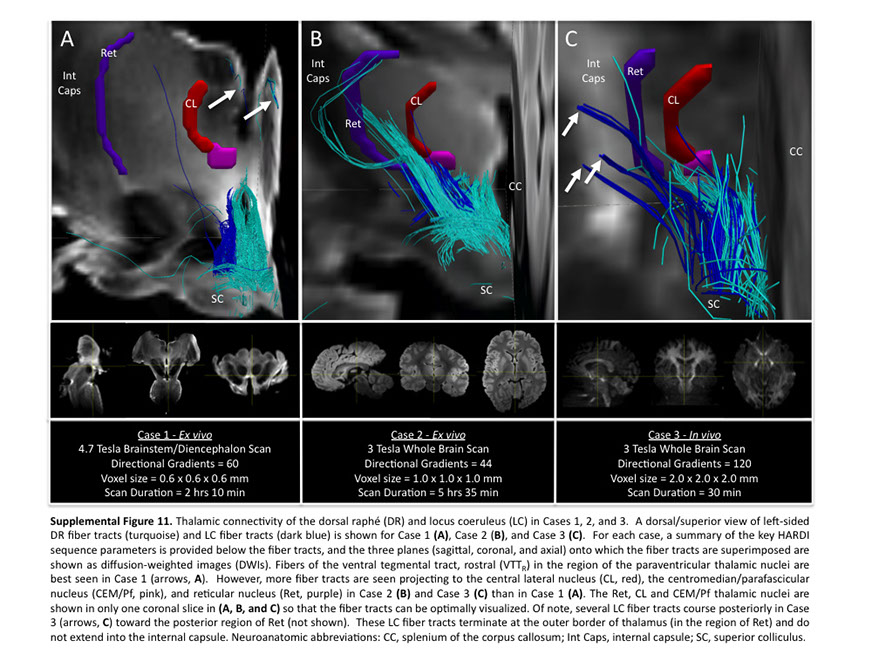
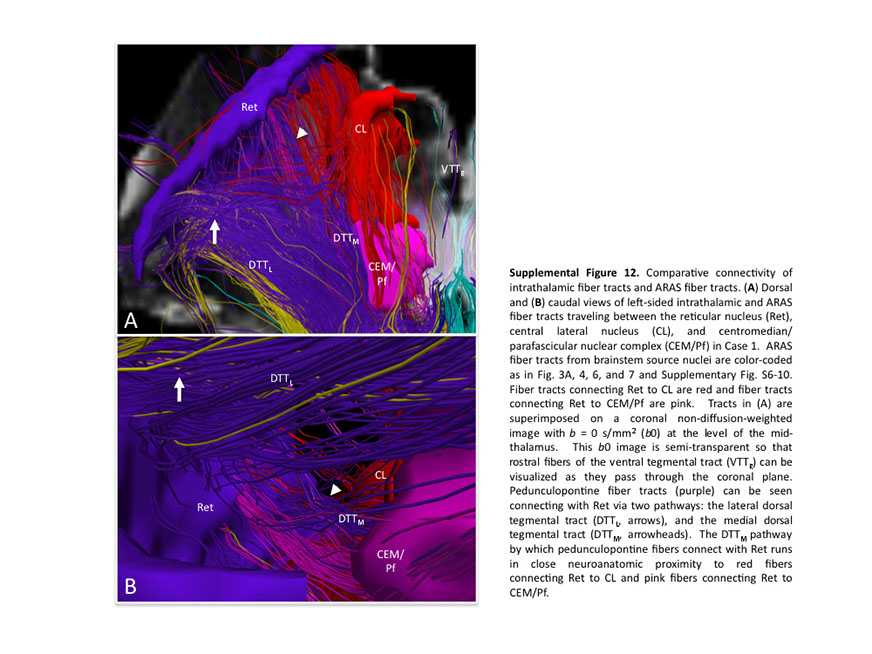
Fatigue diagrams
click for full size, click 'x' button to close 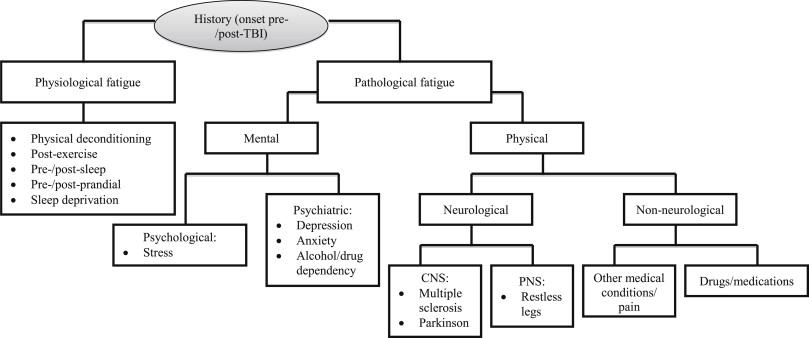
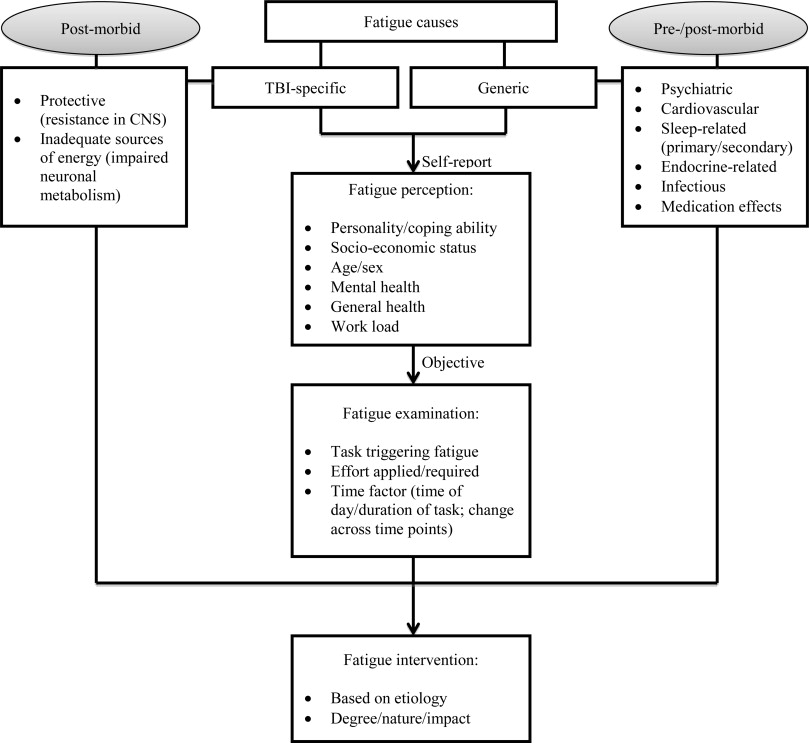 <>xclick here to close and return to report
<>xclick here to close and return to report![]()
1) Hibernation
Hydrogen Sulfide Induces Hibernation State in Chronic Fatigue Syndrome (click to open) ***
Towards understanding the neural origins of hibernation ***
Takeaways from 'Towards understanding the neural origins of hibernation' ***

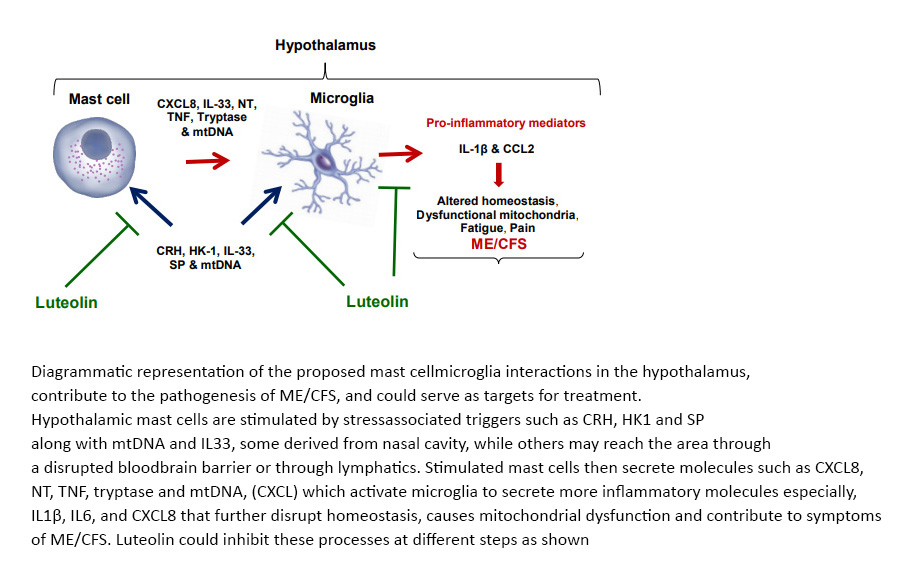
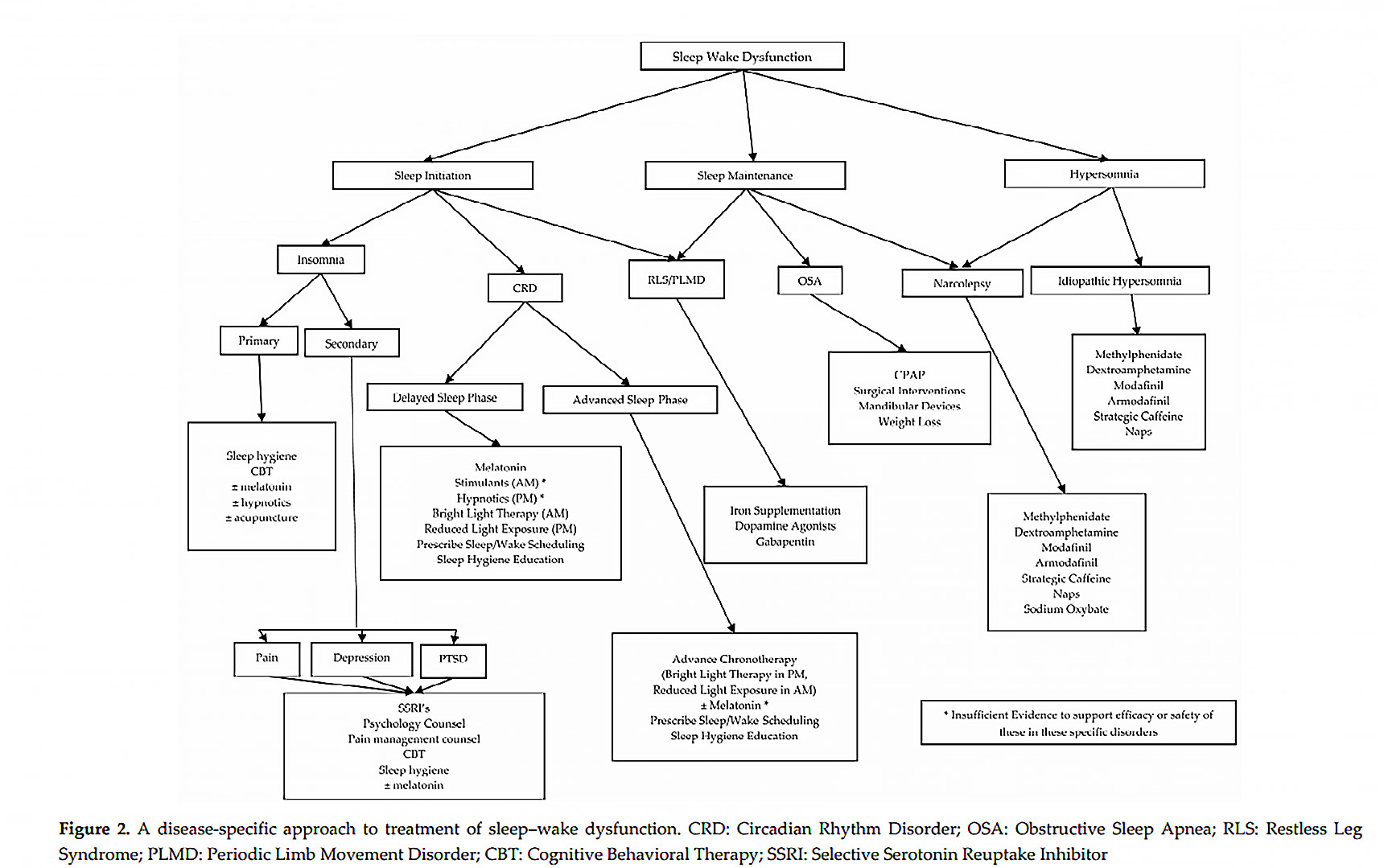
<
>
x
click here to close and return to report

page 32 CFS > ALLOPATHIC MEDICINES > SLEEP, SLEEP DISORDERS, APNEA MACHINE AND SNORE SURGERY
page 31
page 33
back to top
back to top
back to top
back to top
back to top
back to top
back to top
back to top
Click 1
Click 2
Click 3











