Page Synopsis: CFS patients have circulatory issues such as decreased blood volume and cardiovascular preload issue, which is why Pentoxifyline may help. It seems like a rather heavy duty medicine and a commitment (in terms of side effects and monitoring
This page discusses Pentoxifyline as well as cardio related topics
Skill Level 5
Relevance:5 Technical Level:5
page 62 PTBICF > ALLOPATHIC MEDICINE > PENTOXIFYLINE
page 61
page 62b
Please view 'Circulatory Impairment, Disease and Irregularities' section at the full report https://bra.in/7v2NY6** where you'll learn that CFS patients exhibit reduced circulation to brain and other organs (which could explain virtually all symptoms), blood perfusion in the brain, especially in the brainstem, has been shown to be impaired in CFS/ME., cardiac and circulatory problems such as tachycardia and palpitations are common. The usual pattern is high pulse combined with a low blood pressure. Nearly all patients suffer from orthostatic hypotension.
Hypovolemia (low blood volume), Orthostatic hypotension or other dysautonomic states, Hypercoagulation, poor erythrocyte deformability and combinations of these. Reduced circulation to brain and other organs could explain virtually all symptoms. Blood perfusion in the brain, especially in the brainstem, has been shown to be impaired in CFS/ME.8
A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ß2-adrenergic receptors
https://www.sciencedirect.com/science/article/pii/S1568997220300823
Most theories about the cause of symptoms (such as cardiomyopathy and hypercoagulation) do implicate an infectious agent
![]()
https://livingwithchronicfatiguesyndrome.wordpress.com/2011/09/05/pentoxifylline-for-me
Pentoxifylline has the most common brand name, Trental. It is a prescription drug that belongs to the class of drugs called xanthine derivatives. Pentoxifylline’s most common use is to enhance peripheral and cerebral blood circulation.
Mechanisms of Action in ME
Many studies attest to Pentoxifylline’s ability to treat Raynaud’s phenomenon. It achieves improved circulation in vivo through several mechanisms including by improving the flexibility of red blood cells which in turn allows the red blood cells to navigate with greater ease through the capillaries. Pentoxifylline also improves platelet deaggregation which subsequently aids circulation through blood vessels. In addition it reduces the viscosity of blood. As a consequence of the aforementioned mechanisms of action of Pentoxifylline, it is useful in providing oxygen through the means of circulation to the peripheries of the body. SPECT scans have demonstrated that ME patients invariably have impaired blood circulation.
Pentoxifylline has several other possible mechanisms of action in ME other than enhancing circulation. It has the ability to reduce the cytokine IL-2 which is often high in ME patients. Dr. Klimas has stated that “In CFS, the Th2 is upregulated with pro inflammatory TNF-alpha and IL-2.” In Osler’s Web, Hillary Johnson writes, “Abnormally high levels of IL-2 have consistently been found in CFS patients by researchers. The IL-2 levels exceed those found in MS patients, AIDS patients and Lymphoma patients.”
Pentoxifylline may also reduce NF Kappa B which is also high in ME patients. Maes et al. 2007 (http://www.ncbi.nlm.nih.gov/pubmed/17693979) have written that “stimulated production of NF Kappa Beta were significantly higher in CFS patients than in controls. There were significant and positive correlations between the production of NF Kappa Beta and the severity of illness…..It is suggested that CFS patients should be treated with antioxidants, which inhibit the production of NF Kappa Beta.”
The final inflammatory pathway that Pentoxifylline may downregulate is the cytokine TNF-alpha. Moss et al. 1999 (http://www.ncbi.nlm.nih.gov/pubmed/10535608) concluded that “our results suggest a significant increase serum TNF-alpha in patients with CFS compared to non-CFS controls….the clinical testing of TNF-alpha blockers and other anti-inflammatory agents for the treatment of this disease is warranted.”
Pentoxifylline also has a degree of anti-viral activity with Amvros’eva et al. 1993 (http://www.ncbi.nlm.nih.gov/pubmed/8284924) demonstrating its efficacy against 8 viruses. Pentoxifylline was most effective against the herpes simplex virus including a strain that acyclovir was ineffective against, as well as the vaccinia virus, rotavirus and tick-borne encephalitis virus. The authors concluded that Pentoxifylline was an “effective broad spectrum virus inhibitor.”
Pentoxifylline also has the potential ability to inhibit retroviral infection. Several papers have shown that it can (in vitro) inhibit HIV replication and a further study has revealed that Pentoxifylline can reduce the HIV plasma levels in asymptomatic patients. It is sometimes used as a secondary treatment for AIDS.
Dosage
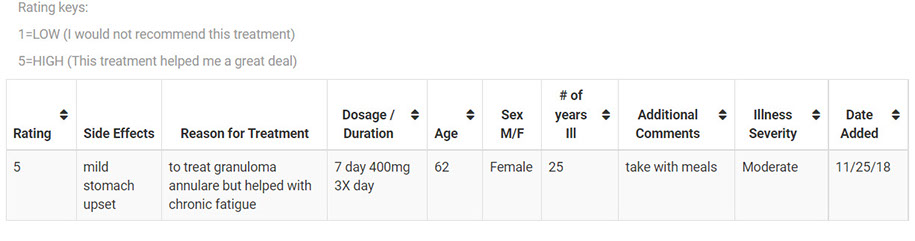
The most common dosage of Pentoxifylline (for diseases other than ME, due to a lack of ME-Pentoxifylline studies) is 400mg taken three times a day at regular intervals for a total daily dosage of 1200mg. Studies have shown that doses of Pentoxifylline at higher than 400mg 3xday do not improve its efficacy however increase the risk of side effects. Due to many ME patients having sensitivities to drugs, a lower initial dosage may be beneficial to reduce the severity of any potential side effects. It is recommended by several sources that Pentoxifylline should be taken at 8 hour intervals in order to maintain a consistent level of the drug in the bloodstream. These sources also recommended that Pentoxifylline is taken with food.
Side Effects and Contraindications
Pentoxifylline is a xanthine derivative (in some respects similar to caffeine) and therefore should not be taken by those patients who cannot tolerate stimulants. Pentoxifylline should also be avoided by those with a peptic ulcer or those with a risk of haemorrhaging. Dr Claudia Zein, a Pentoxifylline researcher has stated that the drug has an “established safety profile” although this comment was not in relation to ME as Pentoxifylline has not been trialled on ME patients in a published study to date. Many studies have tested Pentoxifylline’s safety profile for a variety of conditions with all studies read by me having a similar conclusion. An example of these studies is by Crowder et al. (http://ang.sagepub.com/content/40/9/795.short) who administered the drug to geriatric patients and concluded that Pentoxifylline was “safe and well tolerated” at the 1200mg per day dose.
Drugs.com lists the incidence of side effects of Pentoxifylline http://www.drugs.com/sfx/pentoxifylline-side-effects.html (see extended release capsules, as these are the type commercially available.) In terms of Pentoxifylline and ME patients, a potential negative involves its potential to replicate the cytomegalovirus which some ME patients may have.
Conclusion
Pentoxifylline is a potentially useful drug in the treatment of ME due to its ability to improve cerebral and peripheral circulation. Its anti-inflammatory effects, achieved through down regulating IL-2, NF Kappa B and TNF-alpha may be beneficial to ME patients. Pentoxifylline also has a degree of anti-viral and immunomodulatory ability
User Denise 'Interesting post. My sons (have ME, POTS, joint hypermobility syndrome, as well as Raynauds,) tried pentoxifylline for an extended period of time. One of the reasons for trying it was that it might improve brain blood flow because it reduces the viscosity of the blood.
It did not help my sons but please note that very few medicines/treatments have helped them.
In the USA, pentoxifylline is available only by prescription'
![]()
https://ammes.org/treatment/pentoxifylline
Pentoxifylline (Trental) is a hemorrheologic agent derived from xanthine. A number of stimulants, including caffeine, are derived from xanthine. It improves capillary blood flow by increasing the flexibility of red blood cells and lowering blood viscosity. This double action enables blood cells to squeeze through tiny capillaries with greater ease. Traditionally, pentoxifylline is used to treat vascular diseases, especially those that inhibit blood flow. Pentoxifylline is also used to treat the nausea and headaches associated with altitude sickness.
USES IN ME/CFS: Some clinicians have recommended pentoxifylline in ME/CFS to increase blood flow to the brain. Single-photon emission computed tomography (SPECT) scans consistently reveal lowered blood flow in patients with ME/CFS. In theory, pentoxifylline should resolve that problem.
Pentoxifylline may also address another blood problem common to ME/CFS. The late Dr. L.O. Simpson, a pathologist from the University of Otago Medical School in Dunedin, New Zealand, discovered that patients with ME/CFS had irregularly shaped red blood cells, making it more difficult for blood cells to pass through capillaries. Dr. Simpson believed the decreased cerebral blood flow in ME/CFS was the consequence of abnormally shaped blood cells. Many of the symptoms of inadequate blood supply such as light-headedness, vertigo, and cognitive problems might be alleviated if blood flow were increased.
Apart from pentoxifylline's well-known effects on blood flow, it also has broad antiviral activity. A study conducted in 1993 by a team of Russian scientists found that Trental [pentoxifylline] was an “effective broad spectrum virus inhibitor.” Pentoxifylline also has the ability to inhibit proinflammatory cytokines, which researchers have found to be upregulated in ME/CFS patients
https://www.drugs.com/mtm/pentoxifylline.html
Pentoxifylline causes changes in your blood that help improve blood flow. This also helps your blood carry oxygen to your tissues and organs.
![]()
Pentoxifylline is used to improve blood flow and reduce certain symptoms of a condition called intermittent claudication (IN-ter-MIT-ent KLOD-ih-KAY-tion). Pentoxifylline is not a cure for this condition.
How to take Pentoxifylline and indications (click to open) click again to closePentoxifylline may also be used for purposes not listed in this medication guide. WarningsYou should not use pentoxifylline if you have recently had any type of bleeding in your brain or the retina of your eye. Before taking this medicineYou should not use this medicine if you are allergic to pentoxifylline, or if you are allergic to caffeine or theophylline (Elixophyllin, Theo-24, Theo-Dur, Slo-Bid, Theochron, Theolair, Uniphyl, and others). You also should not use pentoxifylline if you have recently had any type of bleeding in your brain or the retina of your eyes. To make sure pentoxifylline is safe for you, tell your doctor if you have:
How should I take pentoxifylline?Follow all directions on your prescription label. Do not take pentoxifylline in larger or smaller amounts or for longer than recommended. Pentoxifylline is usually taken 3 times each day, with meals. Follow your doctor's instructions. While using pentoxifylline, you may need frequent blood tests. Do not crush, chew, or break an extended-release tablet. Swallow it whole. It may take up to 4 weeks before your symptoms improve. Keep using the medicine as directed and tell your doctor if your symptoms do not improve after 8 weeks of treatment. Store at room temperature away from moisture, heat, and light. What happens if I miss a dose?Take the missed dose as soon as you remember. Skip the missed dose if it is almost time for your next scheduled dose. Do not take extra medicine to make up the missed dose. What happens if I overdose?Seek emergency medical attention or call the Poison Help line at 1-800-222-1222. Overdose symptoms may include severe drowsiness, agitation, fever, flushing (warmth, redness, or tingly feeling), fainting, or seizure. What should I avoid while taking pentoxifylline?Follow your doctor's instructions about any restrictions on food, beverages, or activity. Pentoxifylline side effectsStop taking pentoxifylline and get emergency medical help if you have any of these signs of an allergic reaction: hives; difficult breathing; swelling of your face, lips, tongue, or throat. Call your doctor at once if you have:chest pain;pounding heartbeats or fluttering in your chest;red or pink urine;a light-headed feeling, like you might pass out; orsigns of stomach bleeding--bloody or tarry stools, coughing up blood or vomit that looks like coffee grounds.Common side effects may include:dizziness, headache;nausea, vomiting;diarrhea, gas; orbloating, upset stomach. This is not a complete list of side effects and others may occur. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. Pentoxifylline side effects (more detail)Pentoxifylline dosing informationUsual Adult Dose for Intermittent Claudication: 400 mg orally 3 times a day. If adverse effects develop, reducing the dose to 400 mg twice a day is recommended
bookmarks guy's theory

click here to download the page below
Pentoxifylline Neuroprotective Effects Are Possibly Related to Its Anti-Inflammatory and TNF-Alpha Inhibitory Properties, in the 6-OHDA Model of Parkinson’s Disease https://www.hindawi.com/journals/pd/2015/108179
The pathologic hallmark of PD is the loss of dopaminergic innervation in the striatum and the subsequent degeneration of dopaminergic neurons from SNpc, even though the degenerative process extends beyond that [18]. The pharmacologic treatment of PD can be divided into neuroprotective and symptomatic therapies. However, nearly all available treatments are symptomatic in nature and do not appear to slow or reverse the natural course of the disease. Since there are limited options for PD treatment, neuroprotective agents are currently being tested as means to slow down the disease progression [19].
In the present work, we studied the possible neuroprotective action of pentoxifylline. Our results showed behavioral alterations, in untreated 6-OHDA-lesioned rats, manifested by increased rotations after the apomorphine injection. This circling behavior is a consequence of neuron dopaminergic loss and was attenuated by PTX treatments, suggesting a neuroprotective effect. Furthermore, PTX treatments partly reversed the decreased locomotor activity, as related to untreated lesioned animals. The motor manifestations of PD are attributable to the degeneration of dopaminergic neurons within SNpc, resulting in DA depletion and derangements of neuronal circuits in the target regions of these neurons [20].
In addition, PTX also reversed the increased immobility time observed in the lesioned animals, suggesting an antidepressant-like effect. Others [21] showed that PTX reversed the depressive behavior, demonstrated in a model of myocardial infarction in rats. Depression is one of the most common and persistent nonmotor syndromes affecting roughly 40% of PD patients [5, 22]. Clinically significant symptoms of depression often emerge, before the motor symptoms, persisting throughout the course of the disease [23]. The striatal distribution of adenosine receptors and the antagonistic molecular and behavioral interactions between adenosine and dopamine receptors provide a strong basis for the clinical observation that receptor antagonists enhance motor activity in PD [24]. Interestingly, a clinical study showed that TNF-alpha antagonism may improve depressive symptoms in patients with high baseline inflammatory biomarkers [25].
A striking feature of PD is the preferential loss of DA-producing neurons in the midbrain. It has been proposed that the defective sequestration of DA into vesicles, leading to the generation of reactive oxygen species in the cytoplasm, is a key event in the demise of these neurons in PD and might represent a common pathway, underlying both genetic and sporadic forms of the disease [26]. Thus, despite representing only a symptomatic approach, most of the current therapy for PD focuses on the improvement of the brain DA contents. We showed that PTX, at the higher dose, attenuates the decreased striatal DA levels seen in lesioned animals.
Synaptic degeneration and death of neurons are defining features of PD, while DA-producing neurons in the substantia nigra striatum selectively degenerate [27]. Our results showed a higher fluorescence, indicative of neuron degeneration as demonstrated by the Fluoro-Jade staining in the striatum of the lesioned group, and this feature was greatly attenuated after PTX treatments.
Tyrosine hydroxylase (TH) is the rate-limiting enzyme in brain catecholamine biosynthesis and catalyzes the formation of L-DOPA, the rate-limiting step in the biosynthesis of DA. Thus, PD can be considered as a TH-deficiency syndrome in the striatum [28, 29]. In the present work, the striatal TH immunoreactivity was highly reduced in the ipsilateral side of the lesioned group, and this effect was reversed by PTX dose dependently. Similar results were demonstrated in the immunohistochemical data for the striatal dopamine transporter (DAT).
An important functional role of the dopamine transporter (DAT) is to maintain synaptic DA levels relatively constant and to preserve DA in nerve terminals. A decrease in DAT, despite potentially serving as a compensatory mechanism in early disease, may ultimately result in increased DA turnover and higher oscillations in synaptic DA concentrations, thereby possibly predisposing towards the occurrence of motor complications, as the disease progresses [30]. Thus, PTX effects on these two PD biomarkers, TH and DAT, make this drug potentially useful for PD treatment.
It is largely accepted that there is extensive communication between the immune system and the CNS and that acute or chronic neuron degeneration is associated with microglia activation and release of proinflammatory cytokines [31]. Furthermore, chronic neuroinflammation mediated by microglia plays an essential role in the death of dopaminergic neurons in PD [32–36]. We showed that microglia cells become highly reactive in the ipsilateral striatum of the untreated 6-OHDA animals. On the other hand, a much lower reactivity was observed after PTX treatments, indicating a neuroprotective property. receptor antagonists are potential neuroprotective drugs and the attenuation of microglial NO production could contribute to this neuroprotection [37, 38].
The underlying chronic inflammatory state, evident in PD, strongly suggests a role for neuroinflammation in dopaminergic cell death [39]. Thus, studies by Sawada et al., 2006 [40], reported a marked increase of cytokine levels, in the brain and cerebrospinal fluid of PD patients, and a higher density of glial cells that express TNF-alpha, IL-1 beta, and other cytokines in SNpc of PD patients, as compared to age-matched controls. We showed that while the untreated lesioned animals presented a high immunoreactivity for TNF-alpha, PTX decreased the number of immunopositive cells in the striatum. Evidences [16] indicate that PTX has the potential to inhibit proinflammatory and proapoptotic pathways, via the suppression of TNF-alpha and a caspase-dependent pathway in neuronal PC12 cells, suggesting its protective effects against inflammation-mediated neurodegeneration. Interestingly, recent data indicate that nonmotor features of PD, including depression, are associated with higher CSF levels of inflammatory markers [41].
Apart from the massive loss of dopaminergic neurons, PD brains also show a glial reactivity and neuroinflammation that, besides elevated cytokine levels, are manifested by upregulation of inflammation-associated factors, such as COX-2 and iNOS [17, 42]. In the present study, we clearly showed that PTX treatments of 6-OHDA-lesioned rats decrease immunostainings for COX-2 and iNOS. These data, together with PTX inhibition of TNF-alpha immunostaining, strongly favor the point that PTX neuroprotective action is related to its anti-inflammatory activity.
PTX, besides being a phosphodiesterase inhibitor, increases cAMP levels and decreases TNF-alpha, presenting anti-inflammatory and antioxidant properties, as already shown by us [43] and others [7, 9]. The anti-inflammatory action of PTX is probably related to its ability to suppress oxygen radical production, scavenge reactive oxygen species [44], and blockade extracellular regulated kinase (ERK) phosphorylation and TNF-alpha production [45]. Figure 11 shows sites of possible PTX interventions. A common link in neurodegenerative diseases, including PD, is the presence of neuroinflammatory processes, justifying the importance of anti-inflammatory properties in a potentially neuroprotective drug. Recently [46], we also demonstrated the valproic acid neuroprotection in a PD model. This effect is possibly related to the drug anti-inflammatory action which is, at least partly, the result of its histone deacetylase (HDAC) inhibition. The enzyme HDAC display multiple roles in signaling pathways, and pharmacological modulators of this enzyme possess potent anti-inflammatory and protective effects in several neurological conditions, including neurodegenerative diseases. However, in the particular case of PTX, we believe that the drug anti-inflammatory action is mainly due to its anti-TNF-alpha effect, associated with iNOS and COX-2 inhibitions.
PTX would act by inhibiting steps involved in neuroinflammation and oxidative stress present in PD. Although astrocytes are able to produce TNF-alpha, among other factors, microglia are the major source of this cytokine, during neuroinflammation. Furthermore, INF-gamma is a potent inducer of TNF-alpha gene expression in microglia [12]. BBB = blood brain barrier.
In conclusion, our data demonstrated that PTX exerts a neuroprotective activity in the 6-OHDA model of PD in rats. PTX is also receptor antagonist and these receptors, besides being abundantly expressed within the basal ganglia, are targets to modify abnormal striatal signaling associated with PD [47]. Evidences indicate that receptors signaling pathways mediate the anti-inflammatory effects of PTX [1]. Furthermore, PTX, by increasing the levels of DA, TH, and DAT and decreasing TNF-alpha, appears as a potential candidate to be included in clinical trials, alone or associated with other anti-inflammatory drugs, for the treatment of neurodegenerative pathologies as Parkinson’s disease
Circulatory issues in CFS (click to open) click again to closePlease view 'Circulatory Impairment, Disease and Irregularities' section at the full report
https://bra.in/7v2NY6 White Blood Cells/Leukocytes/Mast Cells/Immune Cells Abnormal Nocturnal Heart Rate Variability Abnormalities in Cerebral Perfusion Blood cells in chronic fatigue syndrome are drained of energy B-Lymphocyte Depletion Capacitance Cardiovascular Preload Issue Cerebral Blood Flow is Decreased in CFS Coagulation Defect Compensated Idiopathic Cardiomyopathy (A Model) Defective vasoconstriction producing POTS in CFS (with cardiac autonomic changes as a secondary response) Dysregulation of Protein Kinase Gene Expression in NK Cells from Chronic Fatigue Syndrome/Myalgic Encephalomyelitis Patients Endothelial Dysfunction Eripheral blood mononuclear cells (PBMCs) Glycolysis High White Blood Cell Count Hypercoagulation Hypoglycemea Hypoperfusion Hypotension Impaired ACh-Mediated Vasodilatation and ACh endothelial sensitivity/Dysfunction Intracranial Compliance (ICC) Lipotoxicity Low Blood Pressure Low Blood Volume (Hypovolemia Low NK Cell Activity NK Cells ERK1/2, MEK1/2 and p38 downstream signalling molecules impaired in CD56 dim CD16+ and CD56 bright CD16 dim Impaired calcium mobilization in natural killer cells (associated with transient receptor potential melastatin 3 ion channels) Loss of Transient Receptor Potential Melastatin 3 ion channel function in natural killer cells from Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients Natural killer cells and single nucleotide polymorphisms of specific ion channels and receptor genes in myalgic encephalomyelitis/chronic fatigue syndrome Lower ambulatory blood pressure Lower Systolic Blood pressure Orthostatic intolerance Oxygen Impaired oxygen delivery to muscle Poor Oxygen Extraction is Contributing to Exercise Intolerance Metabolomics Study Suggests Chronic Fatigue Syndrome May Be Oxidative Stress/Low Oxygen Disease Low oxygen uptake by muscle cells causes exercise intolerance in a majority of CFS patients, indicating insufficient metabolic adaptation to incremental exercise Red blood cells in ME/CFS demonstrate reduced ability to change shape Poor Erythrocyte Deformability Prolonged acetylcholine-induced vasodilatation in the peripheral microcirculation of patients with chronic fatigue syndrome Reduced Absolute Cortical Blood Flow in CFS Reduced Blood Flow short QT interval Small Left Ventricle Stagnant Hypoxia Systemic Exertion IntoleranceAbnormalities in Cerebral PerfusionFrom: A Comparison of Neuroimaging Abnormalities in Multiple Sclerosis, Major Depression and Chronic Fatigue Syndrome (Myalgic Encephalomyelitis): is There a Common Cause?
https://link.springer.com/article/10.1007/s12035-017-0598-z Most of the SPECT studies investigating potential CBF abnormalities in CFS have reported areas of significant cerebral hypoperfusion, at either a global [20, 192] or regional level [193, 194] compared with unaffected controls. However, a minority of researchers reported no significant CBF impairments in comparison with unaffected controls or MDD participants [195, 196]. The reasons for such discrepant findings are unclear, but fewer participants and differences in selection methods may be relevant. The much larger studies conducted by Ichise, Schwartz, Costa and Goldstein, and their colleagues [20, 192,193,194] appear worthy of particular consideration as they examined multiple brain regions and perhaps even more importantly, apart from Ichise and others [192], examined differences in patterns of abnormal perfusion between participants with CFS and MDD, which is currently a contentious area. Ischise et al. compared rCBF in 60 CFS participants and 14 healthy controls and reported significant reductions in cortical/cerebellar rCBF ratios in the frontal, temporal, parietal and occipital lobes, providing objective evidence of widespread functional impairment of the brain in the vast majority of their CFS subjects [192]. Schwartz et al. examined SPECT data differences in terms of regional perfusional deficits and mid-cerebral uptake indices between 45 CFS participants, 14 MDD participants, 27 participants with AIDS dementia complex (ADC) and 38 healthy controls [20]. These authors noted more areas of impaired frontal and temporal perfusion in the ADC group compared with those with CFS or MDD. However, the mid-cerebral uptake index was lower in patients with ADC and CFS than in the MDD and healthy controls groups [20]. The largest study examining potential 99mTc-HMPAO SPECT differences between CFS and MDD patients was carried out by Costa and others [193]. It included 40 healthy participants, 67 CFS participants (both with and without psychiatric co-morbidity) and 29 early- and late-onset MDD participants [193]. These authors reported significantly greater global and brain-stem hypoperfusion in CFS participants free of psychiatric symptoms compared with controls and MDD participants, which was not apparent in CFS participants displaying symptoms of anxiety and depression [193]. Goldstein and others compared high-resolution 99mTc-HMPAO SPECT imaging patterns of rCBF in 33 CFS subjects (55 ± 10 years), 26 age-matched MDD participants and 19 healthy participants in an attempt to investigate the potential pathophysiology of CFS [194]. Importantly, they reported dorsofrontal hypoperfusion as the dominant profile in CFS while hypoperfusion seen in MDD was predominantly in the right orbitofrontal lobe, left temporal lobe and, in particular, the left anterior frontal lobes [194]. More recent research using xenon-computed tomography, ASL and high-resolution SPECT reported consistently impaired CBF at a global and regional level at rest and during exercise [197,198,199,200]. Yoshiuchi and others reported reduced CBF in the right and left hemisphere in CFS participants free of psychiatric comorbidity compared with healthy controls but also noted that hypoperfusion in CFS patients with psychiatric comorbidity appeared to be exclusively limited to the left hemisphere [198]. These results have been broadly replicated by Biswal and others who reported impaired regional and global CBF in the vast majority of their participants with evidence of regional hyperperfusion in a minority [197]. Evidence of hyperperfusion in some CFS subjects has also been supplied by Machale and others who reported significantly increased hyperperfusion in the left thalamus in CFS compared with a pattern of hyperperfusion in the left prefrontal cortex of MDD [200]. Finally, Patrick-Neary and others reported significantly decreased CBF in CFS participants during maximal exercise compared with healthy controls which they suggested as a cause of the profound disability experienced by many patients [199]. The potential capacity of SPECT to differentiate CFS from MDD may be highly significant as CFS is a purely descriptive diagnosis reliant on phenotype only, and there is accumulating evidence that the diagnosis identifies a clinically and aetiologically heterogeneous syndrome rather than a discrete biological entity even when such a diagnosis is made via the application of international guidelines [1, 3, 201, 202]. Unsurprisingly, research in this area has been bedevilled by a lack of reproducibility which is also the case in many other areas where diagnostic categories are descriptive and homogeneity of causation is assumed (reviewed in [203]). This issue in ‘CFS research’ is even more complex because of the frequent use of unvalidated criteria which only mandate the presence of chronic mental and physical tiredness [204] or where a diagnosis is based on the presence of intermittent symptoms based on a population study of fatigue in a localised population [205]. Hence, a method for increasing the homogeneity of a study population and excluding patients with MDD is to be welcomed. The question of heterogeneity is also relevant to the next section of this paper which briefly examines mechanisms whereby systemic inflammation could be a major element initiating and maintaining neuropathology in different illnesses even though investigation with an array of neuroimaging techniques produces very different and often illness-specific results Factors Affecting Severity and Specificity of Neuropathology There is widespread agreement that the relationship between the presence of systemic inflammation and various dimensions of neuropathology is established via the conveyance of inflammatory signals to the brain through a range of humoral and neural routes culminating in the activation of microglia and astrocytes [206, 207]. Once activated, these glial cells secrete a range of inflammatory molecules such as pro-inflammatory cytokines, reactive oxygen species, reactive nitrogen species, prostaglandins and quinolinic acid [208, 209]. These are well-documented effects and have been described in a number of detailed reviews [210, 211].Another source of neuropathology is more indirect however and stems from the loss or corruption of the regulatory functions exerted by these glial cells via a sophisticated communication system with neurones and oligodendrocytes founded on the secretion of exosomes containing mRNAs, miRNAs, caspases and P2X7 (reviewed in [212]). This is a highly complex area, but the key point from the perspective of this paper is that abnormally expressed microglial exosomal miRNAs appear to be independent contributors to the development maintenance and illness-specific patterns of neuroinflammation seen in many different neurodegenerative and neuroprogressive disorders [213]. Thus, microglial activation following prolonged systemic inflammation could produce an illness-specific or at least a range of characteristic WM abnormalities depending on a particular signature of abnormally expressed miRNAs. The mechanisms underpinning this phenomenon are currently unknown but may lie in the recently discovered capacity of at least some miRNAs to activate toll-like receptors and are another mechanism underpinning ‘sterile’ immune activation [214, 215].Microglia also display distinct regional differences in density and transcriptional identity particularly in bioenergetic and immunoregulatory pathways [216, 217]. This underpins observations of region-specific sensitivities to microglial dysfunction and in particular responses to different pathogen-associated molecular patterns [216, 217]. Hence, this provides another mechanism whereby systemic inflammation could produce geographically variable neuropathology. Another highly pertinent example of the role of microglial genetics in creating unique patterns of abnormalities can be found from recent data in MS, where microglia deficient in the production of caspase-8 are considered to be the source of necroptotic markers seen in active plaques of MS patients. These glial cells are likely one of the main drivers of neuronal and oligodendrocyte destruction seen in this illness [218, 219]. The heterogeneous responses of microglia to external or internal stimuli are also under epigenetic regulation involving DNA methylation, histone acetylation and the expression of a wide range of miRNAs [220]. It is also noteworthy that different genomic patterns of DNA methylation within and outside glial cells are associated with different patterns of neuropathology in distinct brain regions [221].Predictably, astrocytes also display extensive variability in biochemical, biophysical and immunoregulatory properties in distinct brain regions (reviewed in [222, 223]). The signalling pathways orchestrated by astrocytes are also strongly influenced by genetics [224]. Importantly, some of the neuroprotective functions of astrocytes are also under epigenetic control. Reactive microglia also compromise astrocytic function and survival in an inflammatory environment with the latter property resulting from a decrease of histone acetylation in astrocytes and a silencing of Nrf-2-mediated antioxidant defences [225]. Antioxidant defence systems also demonstrate pronounced regional differences in composition and efficacy. For example, thioredoxin and thioredoxin reductase levels vary in different brain regions [226]. This is also true of glutathione transferase, glutathione reductase and glutathione peroxidase, with the former two enzymes being reduced in the striatum, neocortex, cerebellum and hippocampus, whereas expression of the last enzyme appears to be reduced in the striatum, cerebellum, cortex and corpus callosum [227, 228]. The expression and activity of NADPH oxidases also displays considerable variation in different brain regions [229]. Selenium distribution across different brain regions is also heterogeneous, with the highest concentrations being in the parietal inferior lobule, putamen and occipital cortex and the lowest levels in the cerebellum and medulla [230]. Different discrete neuronal populations existing in different brain regions display differential susceptibility to neurodegenerative stressors which is a phenomenon often described as selective neuronal vulnerability (SNV). Well-documented examples include neurones in the entorhinal cortex, frontal cortex, hippocampal CA1 region and amygdala, which are the most susceptible to neurodegenerative death in populations of neurones most sensitive to the neurodegeneration associated with Alzheimer’s disease [231, 232]. The increased susceptibility of dopaminergic neurones of the substantia nigra to neurodegenerative destruction in Parkinson’s disease is equally well documented [233, 234]. It should be stressed that the pattern and extent of neural damage seen in different neurodegenerative disease is not just dependent on SNV but also stems from specific factors unique to the aetiology of the illness (reviewed in [235]). SNV also exists within populations in the same brain region. For example, the hippocampal CA1 neurones are much more vulnerable than are CA3 neurones to global cerebral ischaemia [236, 237] and oxidative stress [238, 239].Brain volume and structure are strongly influenced by genetic factors [240,241,242] and resting-state functional connectivity [243, 244], GM and WM volume [245, 246] and CBF [247, 248] are sensitive to environmentally induced epigenetic changes in gene methylation and/or acetylation and/or changes in the expression of a wide range of miRNAs. These factors may also produce heterogeneity in neuroimaging phenotypes between individuals with the same illness or between illnesses even if systemic inflammation is a major driver of neuropathology in each instance CFS is associated with cerebral hypoperfusion, while large VBM studies have shown evidence of GM and WM changes The evidence given in this paper points to the likelihood that systemic inflammation may be a major underlying cause of structural and functional neuroimaging abnormalities reported in seemingly diverse disorders including MS, MDD and CFS Researchers looked specifically at the metabolic processes of oxidative phosphorylation https://en.wikipedia.org/wiki/Oxidative_phosphorylation and glycolysis https://en.wikipedia.org/wiki/Glycolysis two ways cells break apart chemical fuel to transfer energy in respiration. White blood cells taken from 52 patients with CFS and 35 controls were put through their paces under optimal and stressful conditions, testing their capacity to deal with low oxygen levels. There appeared to be a number of key differences in their metabolic processes. But none were as dramatic as the contrast in maximum levels of respiration. By forcing the cells to boost their energy production, the researchers found those with CFS could only squeeze about another 50 percent from their cells – unlike the controls, who nearly doubled their output. "The CFS cells couldn't produce as much energy as the control cells," says Tomas http://www.meassociation.org.uk/2017/11/new-scientist-blood-cells-in-chronic-fatigue-syndrome-are-drained-of-energy-04-november-2017 "At baseline, they didn't perform as well, but the maximum they could reach under any conditions was so much lower than the controls." While the research focused on just one type of cell, it is an important step towards establishing a link between the symptoms of muscle pain, lethargy, and impeded cognitive functions and a biochemical process For more on consistent abnormalities found in CFS patients visit https://bra.in/8pK4xB
![]()
What is meldonium and why was it banned
https://www.wionews.com/sports/what-is-meldonium-and-why-was-it-banned-33353
Meldonium is used to treat ischaemia: A lack of blood flow to parts of the body, particularly in cases of angina, cardiomyopathy and other cardiovascular disorders.The drug also helps to adjust the body's use of energy and can boost stamina and endurance. It increases blood flow, which improves exercise capacity in athletes and increased blood flow means more oxygen to muscle tissue













