Page Synopsis: Allopathic therapies which I'm most interested are:
- Hormone Replacement Therapy (HRT);
- Peptidergic Therapy;
- HBOT;
- Pentoxifylline;
- Troxerutin flavonoid and Cerebroprotein hydrolysate(TCH;
- Donepizil;
- Stem Cell and Stem Cell Precursor
for full list please see personal plan
This page describes the effects of TBI as well as potential therapeutic strategies. It's rather complicated and technical, so you may proceed to the next pages that describe the medical therapies I'm most interested in pursuing
Skill Level 5
Relevance:3 Technical Level:5
page 61 PTBICF > ALLOPATHIC MEDICINE
page 60
page 62
This page presents possible treatments, while the most relevant therapeutics have their own pages
view https://bra.in/5p7mJJ for clickable links, preview below and https://bra.in/8vmgJJ
![]()
'The Injured Brain: TBI, mTBI, the Immune System, and Infection: Connecting the Dots'
Adoptive Immune Therapy to Enhance Microglial-directed Neuronal Repair and Maintenance
Brain injury or trauma causes immune system suppression. Infection is a serious consequence of these events and is present in both open and closed TBI, mTBI, and with stroke. CD4 T-lymphocyte count may be a marker to determine emergence of infection after brain trauma, including stroke. Nosocomial or some HAIs in closed brain trauma (mTBI and stroke) could likely be due to bacterial translocation from the gut and are increased in surgery, trauma, stroke, mucosal barrier problems, or immune suppression of various origins. Bacterial translocation from the gut is decreased with donor and autologous white cell reinfusions. The resultant pulmonary infections from TBI and immune suppression are ameliorated by pharmacologic beta blockade, but not the sepsis. Sepsis and pneumonia are both ameliorated by previously stored autologous white cell reinfusion. Adaptive immune therapy is suggested as a potential therapy for nosocomial or HAIs and in brain trauma infections
![]()
Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics
https://pubmed.ncbi.nlm.nih.gov/27383839
novel preclinical therapies that may have major translational implications. These range from pharmacological agents that are antiinflammatory, cell cycle inhibitors, agents that augment cAMP, noninvasive approaches such as exercise therapy and transcranial magnetic stimulation, and biologics which include stem cells, peptide therapy, and gene therapy.
A major contributor to secondary injury is neuroinflammation that results from chronic microglial activation (i.e., microgliosis). Anti-inflammatory and cell cycle arrest agents have the potential to attenuate the progression of the secondary injury following TBI. One example is minocycline, a second-generation tetracycline that exhibits neuroprotective and potent anti-inflammatory properties.
Specifically, minocycline inhibits proinflammatory cytokines (suppression of IL-1b and IL-6) (Bye et al. 2007) and inhibits microgliosis, and has been shown to be effective in preventing neuronal apoptosis in models of TBI and spinal cord injury (SCI) (Kumar and Loane 2012).
A double-blinded human clinical trial demonstrated significant improvements in motor recovery with minocycline in the setting of SCI (Casha et al. 2012). An additional and a more recent clinical trial showed that minocycline treatment reduced serum neurofilament levels (i.e., a potential biomarker of SCI) in a subgroup of SCI patients (Kuhle et al. 2015). Although many studies have demonstrated long-term behavioral improvements with minocycline, some have shown only transient recovery in neurological outcome (Bye et al. 2007). Another potent anti-inflammatory compound and potential therapeutic agent for TBI and SCI are the synthetic peroxisome proliferator-activated receptor (PPAR) agonists (Villapol et al.
2012; Mandrekar-Colucci et al. 2013; Yonutas and Sullivan 2013; Gensel and Zhang 2015). Upon activation, PPARs translocate to the nucleus to regulate gene expression.
Some evidence shows that PPAR activation suppresses iNOS and COX2, two proinflammatory mediators. Fenofibrate, a PPARa receptor agonist, reduced inflammation, oxidative stress and cerebral edema, and improved behavior after TBI (Besson et al. 2005; Chen et al. 2007).
PPARc receptor agonists, pioglitazone and rosiglitazone, decreased microglial activation, increased antioxidant and
neuroprotective chaperone proteins (Mn-SOD and HSP27/70) and improved behavioral and histological outcomes
after TBI (Sauerbeck et al. 2011; Thal et al. 2011; Yi et al. 2008, potentially through an IL-4-dependent suppression of microglial activation. This would potentially protect the region surrounding the lesion site, and thus promote tissue repair, neuroregeneration, and reduce the progression of secondary injury. Because microglial and astrocyte proliferation is elevated after TBI, an additional approach is to prevent glial proliferation through cell cycle inhibition.
Even postmitotic cell such as neurons can undergo cell cycle activation following injury, and if uncontrolled can cause neuronal apoptosis (Di Giovanni et al. 2005). Cell cycle inhibitors such as flavopiridol, which exerts its effect through cyclic-dependent kinase (CDK) inhibition, has been shown to be efficacious in reducing TBI-mediated lesion volume as well as improving cognition and sensorimotor recovery (Di Giovanni et al. 2005). On a cellular level, flavopiridol specifically blocked cell cycle activation in neurons, astrocytes, and microglia. An additional cell cycle inhibitor, roscovitine, acts directly on CDKs 1, 2, and 5 and has been demonstrated to reduce microglial activation, neuroinflammation, and neurodegeneration weeks
post injury (Hilton et al. 2008; Kabadi et al. 2012).
Therefore, in addition to minocycline, cell cycle inhibitors may also serve to be effective agents for treating individuals
afflicted with TBI in the clinic.
Another potential therapeutic intervention in the setting of TBI is the use of erythropoietin (EPO) (Presneill et al. 2014; Wang et al. 2015; Nichol et al. 2015). EPO is a secreted glycoprotein that acts as a neuroprotective agent by a variety of cellular and subcellular mechanisms which include suppression of apoptosis and inflammatory, antioxidative stress, proangiogenesis, and increased neurotrophic signaling (i.e., prosurvival) (Peng et al. 2014).
Although EPO has demonstrated neuroprotective efficacy in preclinical animal models of TBI, there is still uncertainty as to whether or not it is a useful strategy in the clinical setting (Peng et al. 2014).
There exists some evidence that statins can serve to protect against TBI through its effect on the microvasculature (Kumar and Loane 2012) by increasing nitric oxide production and reducing vascular inflammation. However, there exists great debate on the long-term effects from statins on cognition and behavior due to recent evidence.
Case studies have shown that statins result in attention deficits, decreased psychomotor speed, reduced neuropsychological
performance, and memory loss (Wagstaff et al 2003; Muldoon et al. 2004; Evans and Golomb 2009; Feldman et al. 2010; Maningat et al. 2011; Padala et al. 2012).
Specifically, preclinical findings demonstrated that long-term atorvastatin treatment (7 months) decreased behavior and cognition, altered hippocampal biochemistry, and reduced the subcellular localization of presynaptic vesicular proteins (syntaxin, synaptophysin) (Schilling et al. 2014). More recently, the US FDA placed warning labels on statins for their potential associated cognitive side effects. Furthermore, because statins inhibit intracellular cholesterol synthesis, these compounds could indirectly disrupt formation of cholesterol-enriched plasmalemmal microdomains, cellular regions essential for neuritogenesis, and axonal growth cone guidance [described in more detail in next section (Head et al. 2013). Due to these conflicting findings, more thorough studies need to be conducted to determine whether or not acute or short-term treatment with statins after injury is an effective therapeutic strategy for alleviating complications associated with SCI and TBI in a clinical setting.
In addition to slowing the neuroinflammatory process and protecting viable neurons surrounding the lesion site, additional interventions that evoke structural and functional neuroplasticity can potentially re-establish neuronal circuits and improve behavioral recovery after TBI. After injury, axonal sprouting and growth play an important role in endogenous brain repair. Increased sprouting has the potential to reduce functional deficits post TBI. One such approach is to augment the cyclic nucleotide cAMP (cyclic adenosine monophosphate), a key molecule that enhances neuronal growth (Ming et al. 1997; Song and Poo 1999; Shewan et al. 2002; Murray et al. 2009; Cai et al. 2001).
Post TBI, there is a significant reduction in cAMP signaling pathways (e.g., decreased CREB phosphorylation) (Atkins et al. 2009, 2007). Increasing cAMP levels by phosphodiesterase (PDE) inhibition induce neuronal sprouting, reorganization
of the neurons in the cortex, and recovery of motor function after injury (MacDonald et al. 2007; Atkins et al. 2007). Therefore, pharmacological agents that increase cAMP, direct adenylyl cyclase activation with forskolin, PDE inhibitors (e.g., rolipram, dipyridamole, BC11-38 (Titus et al. 2013b; Atkins et al. 2013; Vang et al., 2010; Ceyhan et al. 2012,
![]()
SSRIs (selective serotonin reuptake inhibitors such as fluoxetine Wang et al. 2011; Kaminska et al. 2013, or SDRIs (serotonin-dopamine reuptake inhibitors such as UWA-121 Huot et al. 2014 may potentially improve functional recovery post injury.
there still exist very few neuroprotective or neuroregenerative strategies for treating this devastating disorder. Although extensive efforts have been put forth preclinically, there have been few successful outcomes in clinical trials (Kabadi and Faden 2014). Little can be done to alleviate the primary injury or insult; it is the secondary or delayed injury that allows for the opportunity for a therapeutic window to prevent progressive tissue damage and loss of function. In order to improve translation of results from preclinical animal models of TBI to the clinic, researchers must address several shortcomings that include the following: (1) improper understanding of the molecular and cellular mechanisms that contribute to secondary injury, (2) insufficient use of multiple brain injury models to test the broader application of the purported therapy, (3) poor understanding of brain pharmacokinetics, (4) lack of cellular and animal models of secondary injury such as hypoxia and effects from toxic cytokines and neuronal growth inhibitory myelin-associated glycoproteins, and (5) disconnect between therapeutic window of potential therapy and the use of clinically relevant behavioral measurements.
Critical to delaying the advancement of secondary injury is to combat the toxic effects of the neuroinflammatory response while simultaneously promoting neuroprotection and neuroregeneration of surviving neurons.
Biopharmaceuticals (Also Known as Biologics or Biological Medical Products)
In addition to pharmacologic interventions for treating individuals afflicted with TBI, there have been some novel and promising advancements based upon preclinical studies that have focused on the use biologics (e.g., stem cells, peptide therapy, gene therapy (DNA, RNA, microRNA, antagomirs), exogenous growth factors, peptides) to combat complications associated with TBI (Mouhieddine et al. 2014). One example is neural stem cell and mesenchymal stem cell therapy, which focuses on neurorestorative and neuroregenerative potential (Wang et al. 2013; Luan et al. 2013; Mouhieddine et al. 2014; Liao et al. 2015). Although there do exist resident populations of neural stem cells in the subventricular zone adjacent to the lateral ventricles and in the subgranular zone of the hippocampal dentate gyrus, the number of migrating neuroblasts in these regions decreases with age (Taylor et al. 2013), thus placing a greater demand for cell replacement therapy after trauma or in age-related neurodegenerative states.
Combining biologics with existing pharmacological agents may serve as an effective therapeutic strategy with translational application. For example, direct inhibition of RhoA or its down-stream kinase (ROCK), both of which are activated following TBI (Sabirzhanova et al. 2013), increases neurite outgrowth. Mechanistically, these inhibitors work by dephosphorylating cofilin, which when not phosphorylated, binds to the actin cytoskeleton and depolymerizes actin-associated filaments, a subcellular event necessary for neurite advancement and neuroregeneration (Hall and Lalli 2010; Dent et al. 2011; Harrington et al. 2011). Thus, using ROCK inhibitors could be an effective adjuvant to biologics for enhancing neuroregeneration after injury.
Growth factors have also been extensively tested for their potential neuroprotective and neuroregenerative efficacy. For example, vascular endothelial growth factor (VEGF), human fibroblast growth factor 2 (FGF2), and BDNF have been shown to indirectly enhance neuronal survival by supporting transplanted stem cells in diseased and injury models (Rizvanov et al. 2011; Ma et al. 2012; Blaya et al. 2014). VEGF and FGF2 also improved functional outcome in a preclinical model of TBI; however, the combination of these two growth factors had little synergistic benefit (Thau-Zuchman et al. 2012). In other occurrences, nerve growth factor (NGF) reduced brain edema following TBI (Lv et al. 2013) and improved functional outcome by reducing beta-amyloid production after TBI (Tian et al. 2012). Some studies have even showed promising effects in murine system using neural stem cells engineered to over-express BDNF (Ma et al. 2012).
In regards to gene therapy, viral and nonviral-mediated delivery systems are being developed for treating neuronal injury. For example, adeno-associated viral (AAV) vectors are used on a regulator basis to deliver and over-express genes of interest in the brain (Murlidharan et al. 2014).
AAVs are nonenveloped, helper-dependent parvoviruses that contain an icosahedral capsid of approximately 25 nm in diameter and 4.7 kb genome. Naturally, they require adenoviruses and herpes simplex viruses for replication and production of viable AAV particles (Bowles et al. 2006).
Three advancements have been made to AAV that allows them to effective tools for gene transfer:
(1) pseudo-typing AAV vectors by using natural or synthetic AAV capsids (Gao et al. 2002; Rabinowitz et al. 2002; Vandenberghe et al. 2009),
(2) cloning of adenoviral helper genes necessary to generate infectious AAV particles (Xiao et al.1998), and
(3) a better understanding that the 145-bp inverted terminal repeats (or ITRs) are the molecular signature for successful packaging of the candidate transgene into the AAV capsid (Xiao et al. 1997). Because of these three major molecular advancements, AAVs are now recombinant (rAAV) and contain minimal contamination of the wildtype virions. AAV vectors have emerged as attractive tools for re-expressing or over-expressing genes of interest to combat neurodegenerative disorders caused by reduced neuronal subpopulations (e.g., hippocampal neurons, dopaminergic neurons), whether due to genetic disorders or environmental events (toxins, injury, ischemia).
In fact, 5.3 % of global clinical trials involving gene therapy have used AAV vectors, yet only a small portion of these trials are attempting to combat CNS disorders (Murlidharan et al. 2014). In addition to viral vectors, nonviral delivery nanoparticles such as micelles, which are aggregates of surfactant molecules (i.e., lipids) dispersed in an aqueous colloid, are being tested in preclinical model of TBI to deliver DNA intranasally (Das et al. 2014; Harmon et al. 2014). A micelle in an aqueous solution that has the hydrophilic ‘head’ exposed to the solvent, while the hydrophobic tail turns inward toward the center of micelle.
Micelles, polyethyleneimine (PEI)-coated micelles, or other micelle-like nanoparticles can be designed to contain genetic material (DNA or RNA) and are of a very attractive manner for gene therapy due to (1) low to no immunogenicity and (2) their ability to enter the brain via intranasal delivery (Das et al. 2014; Harmon et al. 2014), thus removing the need for direct intracerebral gene or drug delivery.
Noninvasive Interventions
Two types of noninvasive interventions that have potential therapeutic implications for improving recovery after TBI are brain stimulation and physical exercise. Transcranial magnetic stimulation or TMS, in both preclinical and clinical studies, have been efficacious in reducing the extent of injury by evoking neuroplastic changes, ultimately leading to recovery of function and improvements in learning (Villamar et al. 2012). TMS involves the use of an extracranial magnetic coil to induce low-frequency (\1 Hz) repetitive TMS to suppress excitability or high frequency ([1 Hz) or intermittent theta burst TMS to enhance excitability [reviewed in (Cramer et al. 2011)]. In addition to being noninvasive, TMS can be used on selectively defined cortical regions with the guidance of neuroimaging techniques in conjunction with physiological measures, such as motor evoked potentials (Neggers et al. 2004) or patient-mediated exercise (e.g., movement of the first dorsal interosseus muscle to measure corticospinal output) (Kleim et al. 2007). Although some in vitro studies using TMS have shown enhanced activation of BDNFTrkB signaling pathway in neurons (Ma et al. 2013), more work needs to be conducted to elucidate the molecular
mechanisms through which TMS produces beneficial effects.
Membrane/Lipid Rafts and Neuroplasticity:
Therapeutic Implications for Neuroregeneration Brain injury disrupts neuronal networks and results in cognitive impairment (Sharp et al. 2014). Structural and functional neuronal connectivity is created by normal neuronal growth, synaptic maintenance and plasticity; all are dependent upon signal transduction pathways and modulation of the cytoskeletal machinery (actin and microtubule dynamics). Key to evoking neuroplasticity and re-establishment of normal neuronal networks after TBI are (1) re-establishing functional signal transduction pathways in the plasma membrane and (2) modulating the cytoskeletal machinery (i.e., actin and microtubule dynamics). Genetic interventions that target both of these mechanisms (i.e., functional signal transduction and regulation of cytoskeletal dynamics) may enhance the efficacy of existing pharmacological agents for improving motor performance and cognitive function after TBI.
Plasticity is an ongoing, endogenous capacity of the nervous system, in which changes in the afferent input or the efferent needs of a neural circuit results in global reorganization that manifests changes on a molecular, cellular, anatomical, and behavioral level. Although plasticity occurs naturally during neurodevelopment or to maintain homeostasis, it is also critical to compensate for injury in order to re-establish functionality. Plasticity is dependent upon changes in individual neurons, neuronal networks, neurotransmitters and ions, gap junctions, and glial cells. Following TBI, recovery of function involves activation of cellular repair mechanisms and structural and functional cellular plasticity, all of which ultimately contribute to anatomical plasticity and formation of new functional connections. Plasticity can in part be evoked by extracellular signals that bind receptors in the plasma membrane to evoke signal transduction that results in polarized motility of the neuronal plasma membrane.
![]()
Subcellularly, intracellular signals (cAMP), modulation of cytoskeletal dynamics (via Rho GTPases), and tethering of the cytoskeleton to the plasma membrane generate a cellular polarity that promotes neuronal growth and plasticity.
Central to this cellular polarity are membrane/lipid rafts (MLR), plasmalemmal ‘hubs’ enriched in sphingolipids, cholesterol, and scaffolding proteins (Head et al. 2013), which organize and regulate (1) TrkB and NMDAR signaling pathways, (2) adenylyl cyclase 8 (AC8) activity and cAMP production, and (3) cytoskeletal dynamics (i.e., Rho GTPase activity) (Kamiguchi 2006; Ayling et al. 2012; Head et al. 2013). Within MLR, the scaffolding and cholesterol-binding protein caveolin-1 (Cav-1) directly organizes neuronal receptors (Head et al. 2013) and the Rho GTPases RhoA, Cdc42, Rac1 (Del Pozo and Schwartz 2007; Grande-Garcia and del Pozo 2008; de Kreuk et al.2011).
MLR are essential for axonal growth and guidance (Denny 2006; Guirland and Zheng 2007), synapse formation (Zonta and Minichiello 2013), and stabilization (Willmann et al. 2006). Evidence shows that loss or disruption of MLR from neuronal membranes inhibits neuritogenesis (Niethammer et al. 2002; Mandyam et al. 2015).
Moreover, TBI causes a disruption in MLR and decreases receptor localization to MLR (Niesman et al. 2014). MLR and Cav-1 provide a signaling membrane platform that tethers and regulates cytoskeletal components necessary for proper neuronal growth, maintenance, and neuroplasticity.
Recent work from our group shows that neurontargeted over-expression of Cav-1 in adult and aged mice (1) increases MLR and MLR-localized expression of growth-promoting receptors (TrkB), (2) augments structural and functional hippocampal neuroplasticity, and (3) improves hippocampal-dependent contextual fear learning and memory in both adult and aged mice (Mandyam et al. 2015). As such, enhancing Cav-1 and MLR formation, specifically in neurons, may evoke the neuroplastic changes necessary to improve motor and cognitive function after TBI. A major focus area of our current research is to use genetic interventions to enhance Cav-1 expressions (via AAV9) and MLR formation specifically in neurons (using a synapsin promoter) (Head et al. 2011) in order to evoke the brain’s natural capacity to regenerate itself and improve functional recovery after injury. Gene therapies, in combination with pharmacologic agents that activate TrkB, activate adenylyl cyclase-mediated cAMP production (serotonin and dopamine reuptake inhibitors), or regulators of cytoskeletal dynamics (Rho GTPase activity) may prove efficacious in promoting functional and structural plasticity, pruning and refining neuritic growth, and rapidly accelerate motor performance and improve cognitive behavior after brain injury
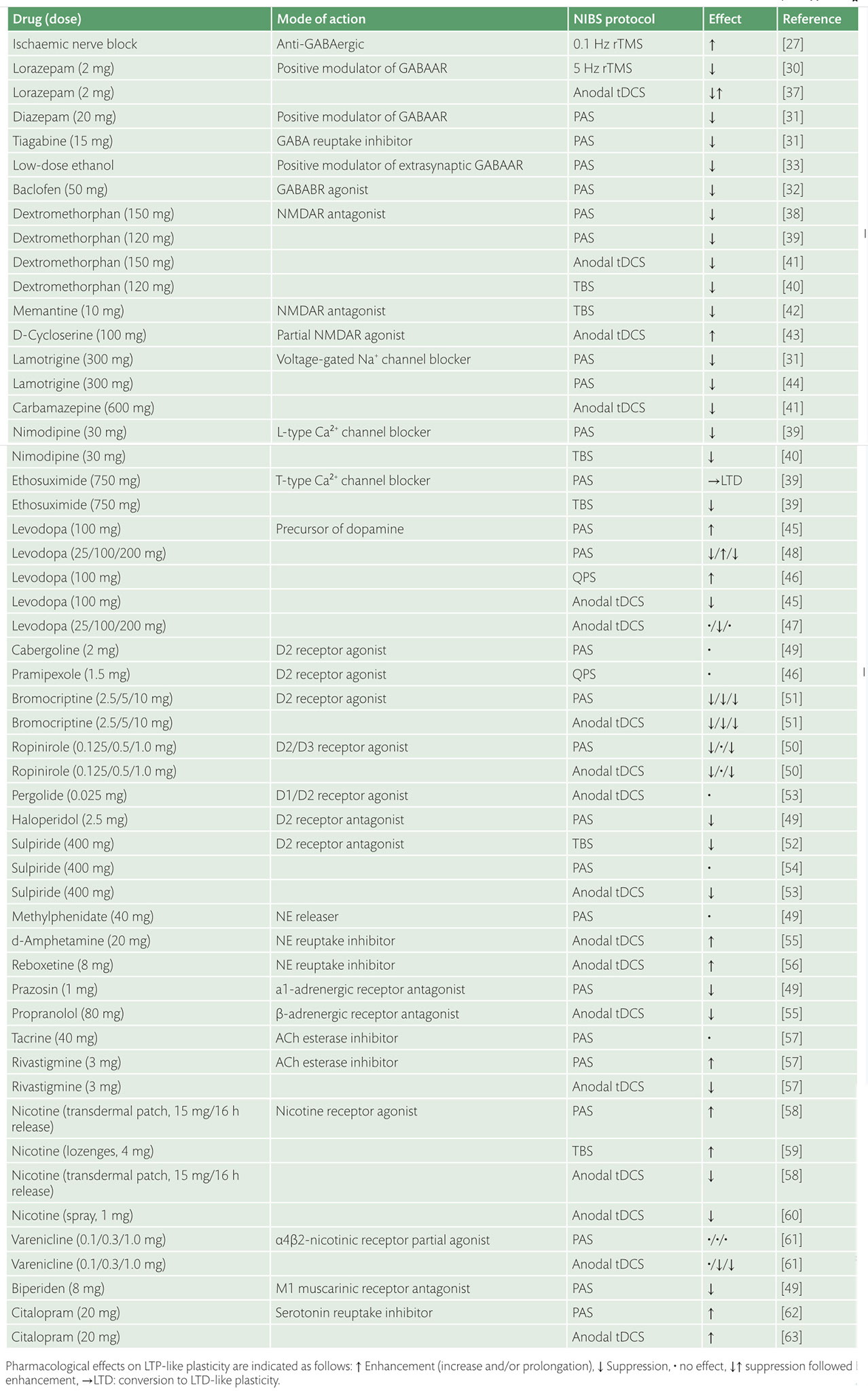
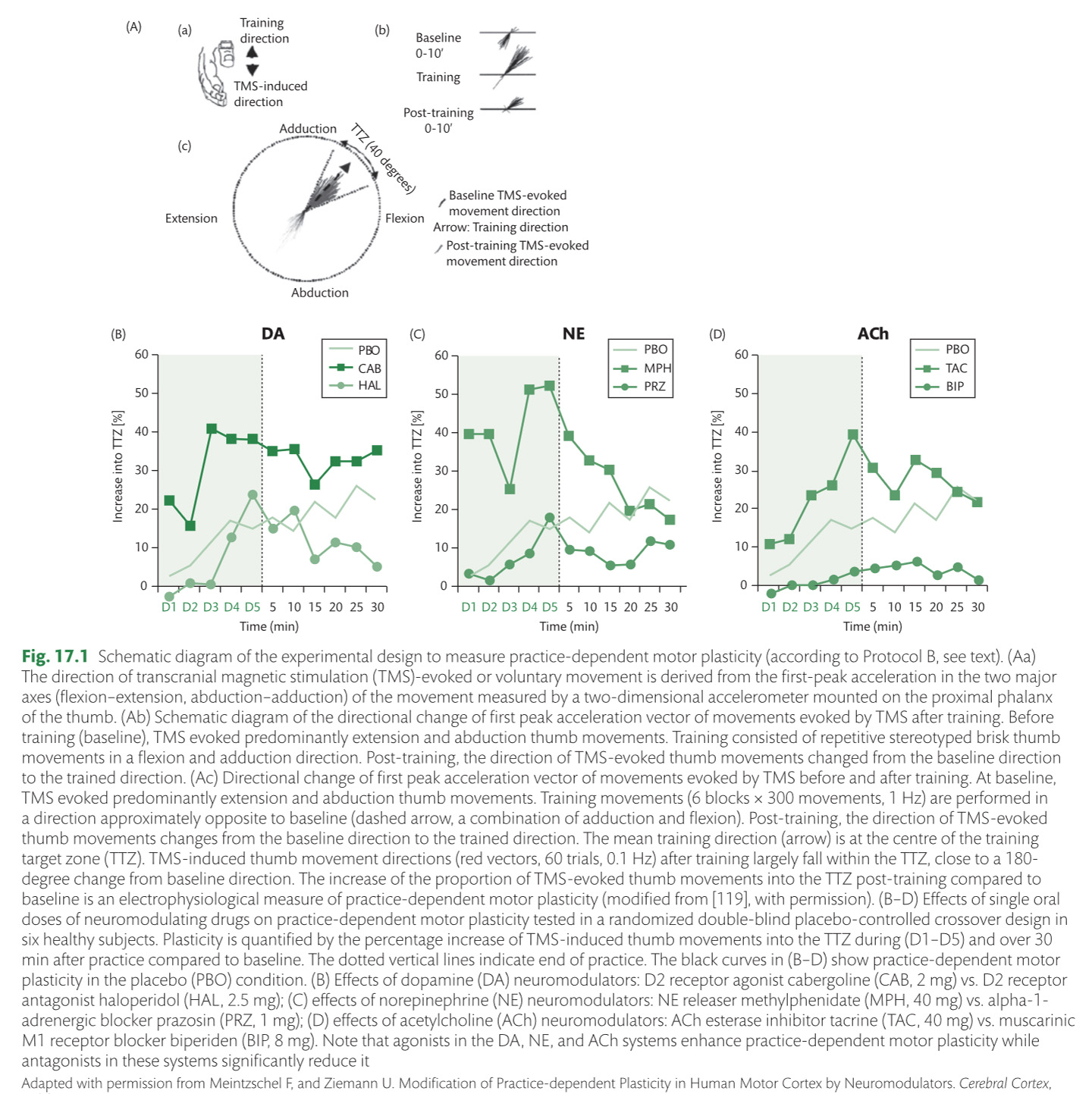
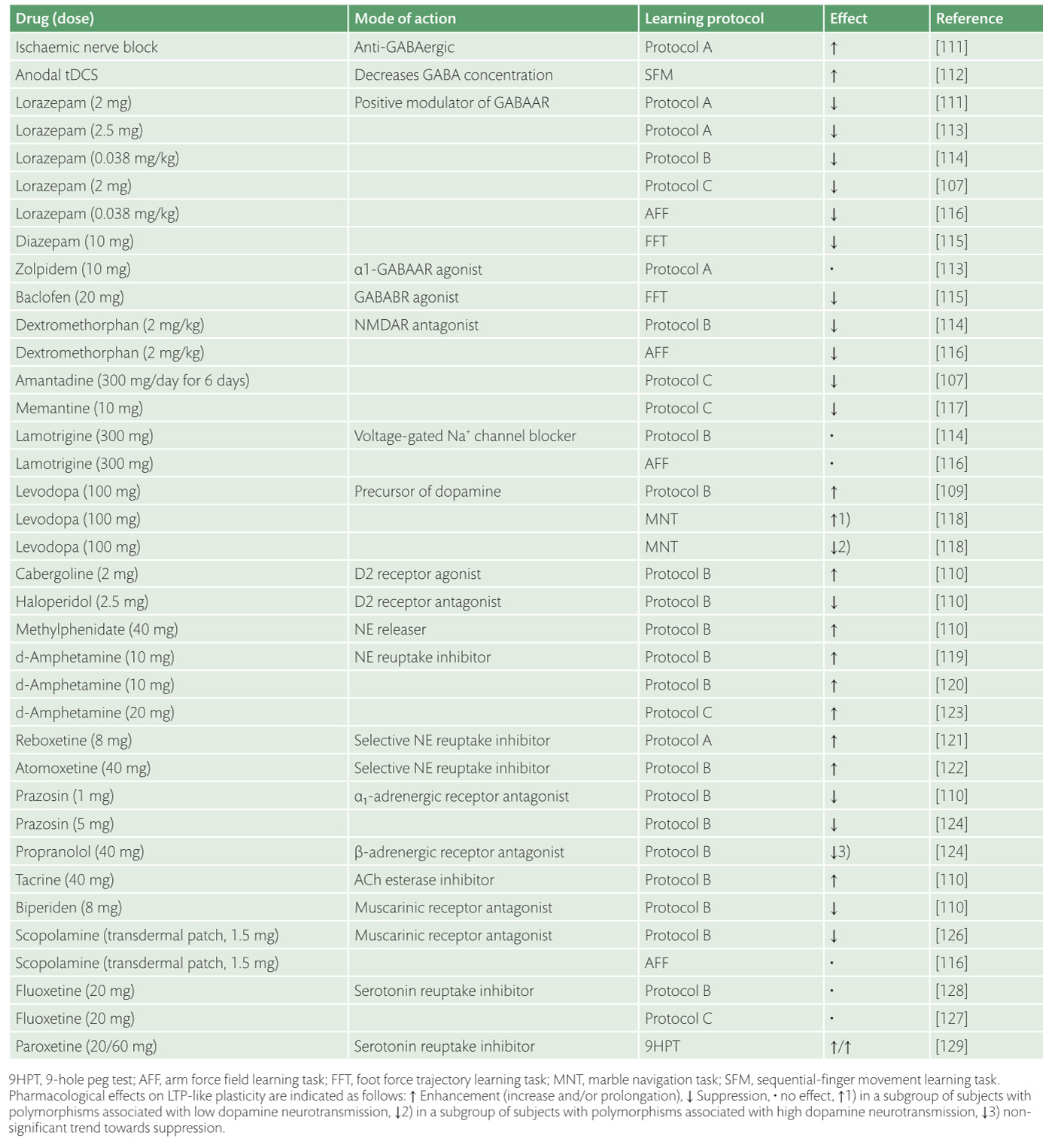
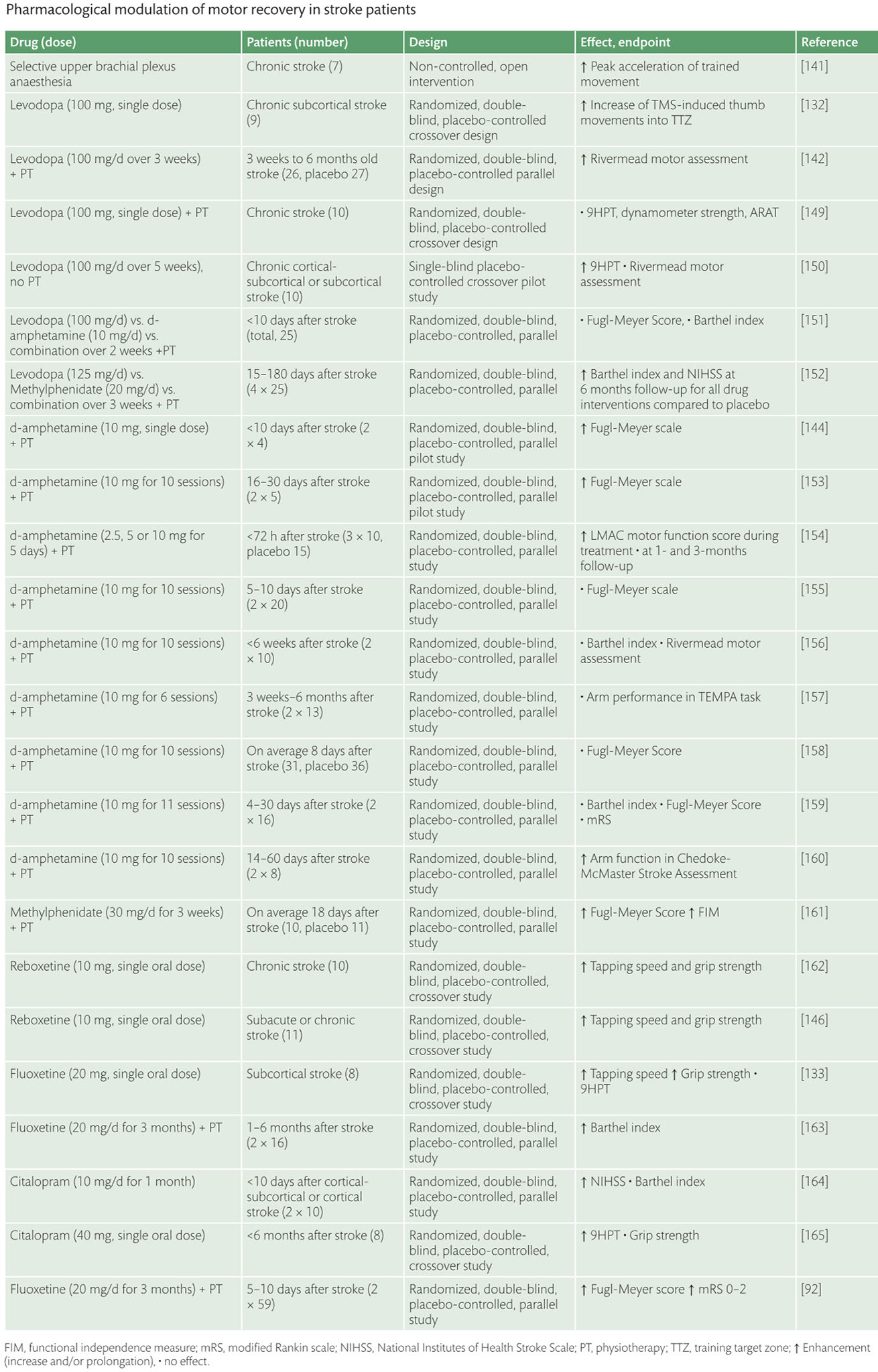
Enhancement of neuroplasticity by drug therapy***
Best Nootropics for Traumatic Brain Injury as PDF, viewed on page 9 or https://nootropicsexpert.com/best-nootropics-for-traumatic-brain-injury

click
<
>
x
click here to close and return to report
Enhancement of neuroplasticity
by drug therapy accompanying tables
click to expand >













