A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ß2-adrenergic receptors
Abstract
Myalgic Encephalomyelitis or Chronic Fatigue Syndrome (CFS/ME) is a complex and severely disabling disease with a prevalence of 0.3% and no approved treatment and therefore a very high medical need. Following an infectious onset patients suffer from severe central and muscle fatigue, chronic pain, cognitive impairment, and immune and autonomic dysfunction. Although the etiology of CFS/ME is not solved yet, there is numerous evidence for an autoantibody mediated dysregulation of the immune and autonomic nervous system.
We found elevated ß2 adrenergic receptor (ß2AdR) and M3 acetylcholine receptor antibodies in a subset of CFS/ME patients. As both ß2AdR and M3 acetylcholine receptor are important vasodilators, we would expect their functional disturbance to result in vasoconstriction and hypoxemia. An impaired circulation and oxygen supply could result in many symptoms of ME/CFS. There are consistent reports of vascular dysfunction in ME/CFS. Muscular and cerebral hypoperfusion has been shown in ME/CFS in various studies and correlated with fatigue. Metabolic changes in ME/CFS are also in line with a concept of hypoxia and ischemia.
Here we try to develop a unifying working concept for the complex pathomechanism of ME/CFS based on the presence of dysfunctional autoantibodies against ß2AdR and M3 acetylcholine receptor and extrapolate it to the pathophvysiology of ME/CFS without an autoimmune pathogenesis.
1. Introduction
ME/CFS is a common and debilitating disorder with a hitherto unknown etiology but of suspected multifactorial origin. A rough recent estimate for the number of patients who may be diagnosed with ME or ME/CFS in the U.S. is 1.7 million to 3.38 million [1]. After decades of neglect and misjudgement as mental disease there is now ample evidence for a complex dysregulation of the immune and autonomic nervous system in ME/CFS. Common “triggers” include severe viral infections and emotional stress. The disease is characterized by severe fatigue and exhaustion, muscular and mental fatigue, exercise intolerance, post-exertional malaise (PEM) and a myriad of symptoms including impaired cognitive ability, poor sleep quality, muscle pain, multi-joint pain without swelling or redness, or headache [2,3]. Many patients suffer from symptoms of autonomic dysfunction including orthostatic intolerance and may complain of dizziness, spatial disorientation, sweating, palpitations or fainting [4]. There is a wide spectrum of non-specific complaints, which may arise secondary to a disturbance that primarily affects the skeletal muscles and includes flu-like symptoms, tender lymph nodes, sore throat, dyspnea and irritable bowel syndrome. PEM, which is a hallmark of ME/CFS, is seen as an aggravation of all symptoms of ME/CFS. PEM involves an abnormal response (e.g., an inappropriate loss of physical and mental stamina, rapid muscular and cognitive fatigability) following physical, cognitive, emotional or orthostatic exertion [[4], [5], [6]].
Bioenergetic muscle dysfunction is also evident in ME/CFS [[7], [8], [9], [10]]. Patients with ME/CFS performed worse than controls in a controlled repeated exercise [11]. A diminished oxygen supply of muscles upon exercise is shown in several studies in ME/CFS patients [12]. In line with this, metabolic changes in ME/CFS indicate hypoxia and ischemia [13]. Skeletal muscle acidosis or dysregulation of protons is found in ME/CFS patients during or after exercise [[14], [15], [16], [17]]. Cultured human skeletal muscle cells from patients with ME/CFS show impaired glucose uptake and ATP levels [18].
1.1. Autoantibodies against ß2-adrenergic receptors and M3 acetylcholine receptors in ME/CFS and their potential pathophysiological relevance
There is ample evidence of immune dysregulation and autoimmunity in ME/CFS [19,20]. Recently, a network of natural antibodies against adrenergic, muscarinergic and other GPCR receptors has been described which are dysregulated in various autoimmune diseases [21]. ß2 adrenergic (ß2AdR) and M3 acetylcholine receptor autoantibodies have been found to be elevated in a subset of ME/CFS patients [22]. Removal of these autoantibodies by IgG apheresis led to rapid improvement in most patients demonstrating a pathophysiological role of autoantibodies in ME/CFS [23]. In a previous study autoantibodies against the muscarinic cholinergic receptor M1 were already reported in ME/CFS patients and were associated with reduced binding of a muscarinic cholinergic receptor ligand in brain detected by PET [24,25].
In support of the idea that autoantibodies against ß2AdR could cause ß2AdR dysfunction and could be involved in the pathophysiology of ME/CFS is the finding that polymorphisms of ß2AdR genes (Gln27 mutation) have been found associated with (adolescent) chronic fatigue syndrome [26]. In healthy adults the Gln27 and the Arg16 mutations were found associated with an unfavorable cardiovascular profile which could be regarded as a mild and asymptomatic form of the cardiovascular situation found in ME/CFS (see below) [[27], [28], [29]]. Analysis of the cardiovascular situation in ME/CFS is the focus of this publication and will be explained in length below.
Numerous studies in ME/CFS showed a decrease heart rate variability (HRV) suggesting a (chronically) high sympathetic tone and a low vagal tone [26,[30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43]]. It is well known from chronic heart failure that adrenergic receptors desensitize by a chronically high sympathetic tone and the ß2AdR is the most sensitive subtype among the different adrenergic receptors for desensitization [44,45]. Altogether, dysfunction of the ß2AdR may be caused by autoantibodies, mutations of the receptor and desensitization. In the presence of a high sympathetic tone as suggested by the HRV studies the association of a high sympathetic tone with ß2AdR dysfunction may lead to severe autonomic dysfunction. The association of both changes cannot be emphasized enough. Could this association explain the enigmatic CV situation of ME/CFS and symptoms?
2. Adrenergic regulation of vascular function
2.1. The role of ß2AdR in sympathetic vascular innervation
ß2AdR are involved in sympathetic vascular innervation which is characterized by a dichotomy: Vasoconstrictor alpha1-adrenergic receptors are opposed by vasodilator β-adrenergic receptors. Whether vasoconstriction or vasodilation occurs strongly depends on the role of vasodilating β-adrenergic receptors versus vasoconstrictor alpha-adrenergic receptors (mainly alpha1-adrenergic) in an organ. In the healthy state sympathetic activation causes vasoconstriction by predominance of alpha1-adrenergic agonism in all organs except for the heart, brain and skeletal muscle. Vasodilation occurs at physiological levels of activation in these organs while a high level of activation as in hemodynamic shock also causes vasoconstriction. Cognitive fatigability and muscular fatigue are hallmarks of ME/CFS. Given the importance of ß2AdR innervation in these organs for perfusion and the possibility that they are dysfunctional the question arises whether fatigue – mental or of skeletal muscles – is causally related to ß2AdR dysfunction causing an issue with perfusion of both organs.
2.2. Evidence for ß2AdR dysfunction in the cardiovascular system in ME/CFS
Is there any evidence for a ß2AdR dysfunction in the cardiovascular system in ME/CFS? Electrophysiological findings in the heart of ME/CFS patients indeed suggest an impaired activation of cardiac ß2AdR supporting the idea that the ß2AdR autoantibodies are dysfunctional by the following findings: Chronotropic incompetence has been reported in ME/CFS during exercise (sinus node effects): Using maximal cardiopulmonary testing (CPET) ME/CFS patients show a blunted rise in heart rate (for review [46]). β1- as well as β2AdR stimulation exert positive chronotropic effects. In the absence of a sick sinus node it is reasonable to assume a dysfunction of ß-adrenergic innervation as ß-adrenergic stimulation mediates physiological tachycardia. Since ß1- as well as ß2AdR stimulation exert positive chronotropic effects the question arises which subtype is dysfunctional. ß1- or ß2AdR or both? A second electrophysiological finding could give a clear answer: Excessive QTc-shortening in ME/CFS (shortened ventricular repolarization) was reported for ME/CFS patients in a situation of orthostatic stress [47]. What are the respective roles of both ß-adrenergic subtypes in ventricular repolarization? ß1-adrenergic receptor stimulation shortens the QT-interval by activating cardiac repolarizing “slowly activating delayed rectifier potassium channels” (IKs) This strongly suggests intact ß1-adrenergic receptor function. ß2AdR stimulation prolongs the QT-interval by inhibiting the cardiac “rapidly activating delayed rectifier potassium channels” (IKr) [48]. Therefore, QTc shortening can be explained only by ß2AdR dysfunction in the presence of intact ß1-adrenergic receptors and a high sympathetic tone.
Table 1 shows location and function of ß2AdR. Vasodilator ß2AdR are found in brain vessels and in the vasculature of skeletal muscles, probably to oppose or limit alpha-adrenergic vasoconstriction or to induce net vasodilation [27,[49], [50], [51]]. Since autoantibodies against the M3-receptor has also been found their location and function should also be briefly mentioned. This receptors is ubiquitously expressed in vascular endothelium to release nitric oxide (NO) [52]. Cholinergic vasodilation is predominantly mediated by the M3-receptor [53]. Both receptors, ß2AdR and M3-receptor can be considered as important vascular vasodilators of the autonomic nervous system with particular importance for cerebral and muscular blood flow [27,54]. Assuming that the reported antibodies against important vascular receptors cause dysfunction of the receptors it is reasonable to consider the possibility that mental and muscular fatigue could be related to a disturbance of blood flow. Due to its ubiquitous expression in endothelial cells M3 acetylcholine receptor dysfunction may mainly affect blood flow in the whole body (endothelial dysfunction). In the organs where the blood vessels are also endowed with ß2AdR the dysfunction of the M3-receptor may act highly synergistically with ß2AdR receptor-dysfunction to impair flow at rest and particularly at activation. Here we focus on the ß2AdR because the ß2AdR is clearly associated with cerebral and skeletal muscle perfusion.
Table 1. Location and function of ß2AdR.
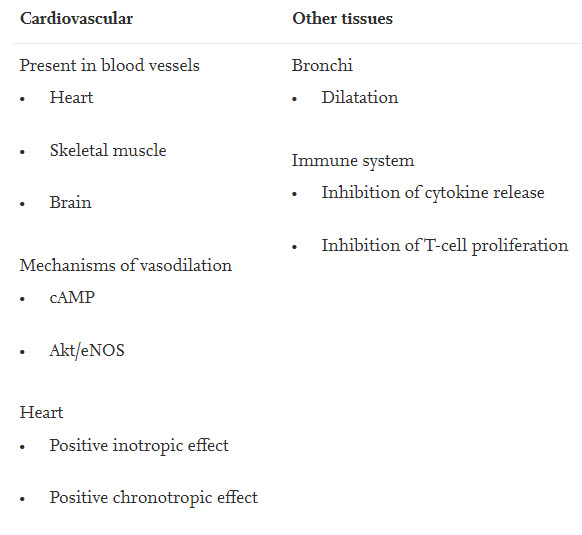
3. Functional sympatholysis in skeletal muscle
The term functional sympatholysis in skeletal muscles describes the mechanisms that prevent that vasoconstrictor alpha1-adrenergic receptors in skeletal muscle during exercise or mental activity cause vasoconstriction. It comprises a number of different mechanisms. The precise mechanisms of functional sympatholysis and exercise-induced vasodilation and muscular hyperemia have not been fully elucidated but what seems clear is that vascular ß2AdR contribute to vasodilation in skeletal muscles in exercise induced hyperemia and functional sympatholysis as do ß1- and ß3-adrenergic receptors [[55], [56], [57]]. If ß2AdR were dysfunctional the vasodilator forces would be weakened. The second most important mechanism of functional sympatholysis is the metabolic stimulation of the release of endogenous, short-lived vasodilators that are released locally and that are supposed to act only locally. These mediators comprise adenosine, ATP, prostaglandins (PGEs), prostacylin (PCI), bradykinin (BK), protons, and others. Algesic vasodilator substances were also found released in heavily working muscle with unlimited perfusion (microdialysis) [58]. These mediators are supposed to be involved in functional hyperemia during exercise where skeletal muscle blood flow can rise by at least 20-fold. These mediators are not only released for the purpose of functional hyperemia they were also found elevated in ischemia in skeletal muscle and in the heart [[59], [60], [61], [62], [63], [64], [65]]. The mediator with the most dramatic actions released in ischemia is the nanopeptide bradykinin released by the kallikrein-kinin-system (KKS) from blood precursors, the kininogens, by the action of kallikrein. Bradykinin (and PGE2 and prostacyclin) causes vasodilation and microvascular leakage. Bradykinin is more potent than histamine in doing this and it is the most potent opener of the blood-brain-barrier (BBB) [66,67]. The mediators released by ischemic muscles or in the process of functional sympatholysis as a response to excessive vasoconstrictor influences are also important pain mediators and are assumed to cause angina pectoris pain in cardiac ischemia, particularly bradykinin and prostaglandins [60]. Bradykinin is a most potent algesic endogenous vasodilator peptide also having hyperalgesic and spasmogenic effects [68,69]. Bradykinin stimulates the production of prostaglandins such as PGE2 which are known to cause pain and hyperalgesia so that even smaller algesic stimuli (other modalities) are perceived as painful. Activation of cyclooxygenase (COX) by bradykinin contributes to many but not all of its effects [68,70,71].
3.1. Does spillover of algesic vasodilators released from skeletal muscles into the systemic circulation occur in ME/CFS?
As autonomic vascular innervation fails in skeletal muscles metabolic regulation could gain importance, which would be strongly favored by a still enigmatic metabolic disturbance in skeletal muscle. Table 2 summarizes possible causes for functional sympatholysis in ME/CFS to oppose (increased) alpha1-adrenergic-induced vasoconstriction.
Table 2. Possible causes for enhanced functional sympatholysis in skeletal muscle in ME/CFS.
1) Dysfunctional ß2AdR due to autoantibodies, mutants and desensitization.
2) High sympathetic (vasoconstrictor) tone as suggested by HRV-studies [[30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44]]. Sympathetic tone may be enhanced as a reaction to dysfunctional ß2AdR. Hypovolemia typically found in ME/CFS enhances sympathetic tone.
3) Endothelial or vascular dysfunction of any cause, which is found in patients [72,73]. Dysfunctional ß2AdR would also cause vascular dysfunction raising the vasoconstrictor efficiency of alpha1-adrenergic stimulation.
4) Poor background bioenergetic situation due to an unexplained metabolic (mitochondrial) disturbance cited above: The signal for this metabolic stimulation of the release of algesic vasodilators is a short fall of ATP in the beginning of exercise which is expected to be strongly enhanced in the presence of a bioenergetic disturbance of the skeletal muscle.
Could this lead to excessive compensatory generation of algesic vasodilators in the large body skeletal mass (e.g. during mental stress) so that high amounts are released? Could spillover of excessively produced algesic vasodilators into the systemic circulation – despite short half-lives - explain the enigmatic CV situation and symptoms of ME/CFS? A paradigm for such a spillover of inflammatory mediators from an ischemic muscle is severe dysmenorrhea where prostaglandins (PGE2) are released from a hypoxic uterus, which is hypercontractile and develops intramuscular pressures >120 mmHg to severely reduce perfusion, into the circulation causing systemic complaints [74]. PGE2 levels are found elevated in blood. Such a spillover into the systemic circulation is considered responsible for symptoms of fatigue, flu-like symptoms, fever, pain and sleep disturbances in dysmenorrhea [74]. PGE2 having awakening effects may explain sleep disturbances in dysmenorrhea [75]. There is a wide overlap of symptoms with ME/CFS. Since the much larger skeletal muscle mass is affected in ME/CFS compared with the ischemic uterus even a moderate disturbance of the bioenergetic muscular situation or of the autonomic vascular regulation could produce relevant concentrations of those algesic endogenous vasodilators which we assume cause systemic effects. Probably due to the similarity of symptoms many women with ME/CFS have a diagnosis of dysmenorrhea or endometriosis as well [76].
4. The cardiovascular situation in ME/CFS
4.1. The unique cardiovascular situation of ME/CFS with hypovolemia, preload failure, small hearts, high sympathetic and low vagal tone and an inhibited RAAS
The cardiovascular situation in ME/CFS patients is unique and not found in any other condition or disease. Low vascular, atrial and ventricular filling suggesting hypovolemia, and preload failure has been found in a number of studies leading to a low stroke volume and a lower than normal cardiac output (CO) at rest while the RAAS activity is low [[77], [78], [79], [80], [81], [82], [83]]. Therefore, ME/CFS is frequently associated with orthostatic dysfunction (OD) and the postural tachycardia syndrome (POTS). Small hearts have been typically reported for ME/CFS. Cardiac hypotrophy may therefore develop chronically as a consequence of chronic low ventricular filling and a low RAAS activity. Endothelial dysfunction has been reported which is expected to increase vasoconstrictor efficiency. HRV indicated a high sympathetic (vasoconstrictor) tone. What could be the cause of the low filling state of the vasculature?
4.2. Preload failure in ME/CFS, a consequence of hypovolemia by microvascular leakage and renal hyperfunction and venous vasodilation/ pooling?
According to the considerations made above endogenous vasodilator substances could be produced in skeletal muscles with a poor bioenergetic situation or for the purpose of functional sympatholysis. Bradykinin for instance is not only a painful and vasodilator mediator it also very potent in increasing endothelial (microvascular) permeability [84,85]. In the systemic circulation this could cause a fluid shift from the intravascular space to the interstitial space.
Apart from reducing cardiac output a diminished filling of the vasculature activates the sympathetic nervous system, reduces vagal tone and it would be expected to activate the renin-angiotensin-aldosterone system (RAAS). Increased (vasoconstrictor) sympathetic tone and reduced vagal activity have indeed been reported in a number of heart rate variability studies in ME/CFS as mentioned above and could be explained by hypovolemia. The real surprise is that the RAAS is not activated in ME/CFS, but renin production and aldosterone are even decreased, in a situation of hypovolemia which is the typical physiological stimulus for its activation [[86], [87], [88]]. The inability to activate the RAAS prevents refilling of the vasculature and leads to a rise in sympathetic tone with its enhanced vasoconstrictor efficiency. Understanding these unique and enigmatic cardiovascular findings of ME/CFS in the myriad of confusing symptoms could be the key to understanding the pathophysiology of ME/CFS.
Bradykinin generated in the ischemic muscle after reaching the systemic circulation may contribute to the low vascular filling state in ME/CFS by different mechanisms to which PGE2 and prostacyclin having similar renal effects may contribute [89]. Increased microvascular leakage is not the only cause of hypovolemia but renal excretory effects of bradykinin and inhibition of renin production could contribute. The potential effects of bradykinin on vascular and renal function are summarized in Table 3.
Table 3. Potential effects of bradykinin related to the vascular filling state.
1) Extravasation of intravascular fluid by increasing endothelial permeability lowering intravascular volume. Vascular leakage induced by bradykinin may be exaggerated by dysfunctional ß2AdR autoantibodies as it has been reported that ß2AdR agonists reduce microvascular leakage induced by bradykinin in experimental settings [84], [90]. Prostaglandins increase the vascular permeability induced by bradykinin [71]. Plasma albumin levels were increased in severe ME/CFS in line with the assumption of enhanced microvascular leakage [91].
2) Renal actions of bradykinin: Bradykinin increases renal blood flow by its renal vasodilator effect and inhibits sodium reabsorption in the distal tubule causing renal hyperexcretion and hyperfunction (acting like a mild salidiuretic drug) [92]. Indeed decreased creatinine and urea levels were found in patients with severe ME/CFS in line with renal hyperemia supporting the idea of excessive renal excretion as a cause of hypovolemia [91].
3) Inhibition of renin production: falling pressure in the afferent renal arteries reduced sodium concentration in the distal tube and ß-adrenergic stimulation (mainly ß1-adrenergic) are the main physiological stimuli for renin production. The renal actions of bradykinin could explain the deficient renin activation and the RAAS paradoxon of ME/CFS as it [96] increases renal blood flow and has natriuretic and diuretic effects to annihilate the signals that otherwise stimulate renin secretion [[89], [92], [93], [94], [95], [96]].
These actions of bradykinin are in line with the general view that the kallikrein-kinin-system (KKS), which releases bradykinin, physiologically opposes the effects of RAAS with regard to its main function of volume regulation [89,[93], [94], [95],97]. It remains to be investigated whether in ME/CFS there is a dysbalance between these two systems favoring the KKS.
There is potential role of heat shock protein90 (Hsp90) in the activation of the KKS: Hsp90 can activate the KKS to release bradykinin independent of FXII and it is released from endothelial cells stimulated by estrogen, IL-1 and TNF [98]. This could be related to both the frequent post-infectious appearance (IL-1 and TNF) and the female gender preference (estrogen). As mentioned earlier different mechanisms are involved simultaneously in functional sympatholysis or hyperemia in skeletal muscles. It seems possible that in ME/CFS the qualitative composition of the vasodilator mechanisms or mediators is switched by Hsp90 towards the release of algesic and inflammatory mediators like bradykinin which are typically released in ischemia.
Infusion studies of bradykinin in humans did not only show a decrease in peripheral resistance indicative of the dilatation of arterial resistance vessels but also a reduced venous tone indicating that bradykinin could lower cardiac preload by increasing venous blood pooling by causing venous dilatation [99]. In the presence of hypovolemia enhanced venous pooling is particularly detrimental.
Spillover of vasodilators released from the muscles into the arterial system during exercise would also be unfavorable for muscle perfusion because it could cause uncoordinated vasodilation in the other organs as explained for the kidney, thereby diverting blood from the muscle (steal-effect). Vasodilation in the intestine may also lead to intestinal hypervolemia at the expense of preload. Table 4 gives an overview of the disturbed hemodynamic situation in ME/CFS.
Table 4. Disturbed hemodynamic situation in ME/CFS: Cardiovascular causes.
A) Presumed mechanisms of preload failure in ME/CFS
•
Hypovolemia
o
Microvascular leakage by bradykinin (unleashed by ß2AdR dysfunction)
o
Renal hyperexcretion caused by bradykinin and prostaglandins
▪
Renal vasodilation
▪
Inhibition of tubular sodium reabsorption
▪
Inhibition of the signals for renin generation, low RAAS activity and low aldosterone
•
Venous pooling by venous dilation and potentially intestinal hypervolemia
B) Presumed mechanisms of muscular and cerebral misery perfusion in ME/CFS
•
Preload failure as explained in A) leading to lower cardiac output
•
Cardiac causes
o
Cardiac hypotrophy (small hearts)
o
Cardiac fibrosis in some patients impairing diastolic filling [100]
o
Chronotropic incompetence and diminished inotropic component (due to dysfunctional ß2AdR)
•
Disturbances of the arterial system
o
Impaired vasodilator activity due to endothelial dysfunction favoring alpha1-adrenergic vasoconstriction
o
Enhanced sympathetic tone and vasoconstriction due to hypovolemia, stress and pain. Vascular remodeling by alpha1-adrenergic receptors (hypertrophy)?
o
Steal effect for muscle perfusion due to spillover of vasodilators causing vasodilation in other organs during exercise
Apart from causing metabolically induced vasodilation by locally released vasodilators (mechanisms of functional hyperemia) relative underperfusion of active skeletal muscles in the early phase of an exercise causes metabolites to accumulate and stimulate skeletal muscle afferents, which triggers the so-called muscle metaboreflex activation (MMA). MMA activation in normal subjects during submaximal dynamic exercise increases arterial pressure (to raise the perfusion pressure) primarily via increases in stroke volume and less by an increase in peripheral vasoconstriction [101]. MMA also mobilizes central blood volume enabling increases in preload and by that in stroke volume. Thus, metabolic regulation of blood flow to the working skeletal muscle has a systemic (MMA) and a local component, the metabolically stimulated release of (algesic) vasodilators. Both should be affected by the poor bioenergetic situation in skeletal muscle in ME/CFS.
In subjects with type 2 diabetes mellitus BP response to the MMA relies more on an increase in systemic vascular resistance than on cardiac output increments, presumably as a consequence of endothelial dysfunction raising vasoconstrictor efficiency [102]. Due to preload failure in ME/CFS patients it seems impossible that MMA could achieve an adequate rise in perfusion pressure by an adequate increase in stroke volume at the onset exercise; the only possibility of raising BP is by vasoconstriction [83]. Concomitant activation of the baroreflex in the upright position, which is strongly activated by hypovolemia in ME/CFS, has the potential to aggravate the situation: in experimental heart failure it has been shown that concomitant activation of the baroreflex and the MMA act synergistically to cause vasoconstriction in skeletal muscles [101]. The situation would be aggravated by a dysfunctional ß2AdR with its vasodilator function in skeletal muscles. MMA activation may be enhanced by the poor bioenergetic situation of the skeletal muscle contributing to vasoconstriction in the particular hemodynamic situation of ME/CFS. Hence, compared with the healthy situation three factors could act in concert to cause excessive vasoconstriction instead of vasodilation with the onset of exercise in the upright position: ß2AdR dysfunction, excessive baroreflex activation due to hypovolemia and excessive MMA due to poor metabolic situation. This may stimulate the postulated release of algesic vasodilators like bradykinin in skeletal muscles as a compensatory vasodilator mechanism which may be effective to a certain extent in a compensatory manner to raise local perfusion but excessive production of these vasodilators could lead to the spillover into the systemic circulation. Various symptoms of ME/CFS could be explained by the spillover of algesic vasodilators as summarized in Table 5.
Table 5. ME/CFS symptoms potentially explained by spillover of (hyper)algesic vasodilators into the circulation.
Pain, hyperalgesia, flu-like symptoms: These could be explained by excessive generation of algesic vasodilators in skeletal muscle and spillover into the systemic circulation. In line with the assumption that hyperalgesic mediators are excessively generated during exercise to enter the systemic circulation pain threshold is decreased in ME/CFS after exercise (hyperalgesia) while it is normally increased in healthy subjects (hypoalgesia) [103]. Quantitative sensory testing demonstrated heat and mechanical hyperalgesia in CFS subjects [104].
Headache may arise from the overproduction of those inflammatory vasodilators. Apart from releasing PGE2 and displaying direct algesic effects bradykinin stimulates the release of substance P and cGRP from sensory nerve fibres (referred to as neurogenic inflammation), recognized mediators of headache [105]. Prostaglandins could contribute to CNS symptoms by fever, headache and by hyperalgesia and by disturbing sleep (awakening effect of PGE2).
Sore lymph nodes: The feeling of sore lymph nodes can be explained by microvascular leakage plus hyperalgesic action leading to painful distension of lymph vessels.
Intestinal and bladder complaints: These can be explained by microvascular leakage (edema) and vasodilation. Intestinal hypervolemia may not only divert blood from skeletal muscles during exercise (steal effect) but contribute to preload failure. Pain in intestinal organs may be explained by the spasmogenic and hyperalgesic actions. Bronchi may be narrowed by by airway edema, dysfunctional ß2AdR and spasmogenic action of bradykinin and prostaglandins [106].
Mental stress: Mental stress may in the presence of vascular dysfunction and a high sympathetic tone may cause vasoconstriction in skeletal muscles leading to the release and spillover of algesic vasodilators to cause the typical symptoms. Passive muscles may be particularly sensitive to vasoconstriction as the powerful mechanisms that lead to functional hyperemia in working muscles are not operative.
Mental fatigue and cognitive problems (brain fog): These may arise from reduced cerebral blood flow (CBF) by excessive sympathetic vasoconstriction in the presence of dysfunctional ß2AdR and vascular or endothelial dysfunction. ß-adrenergic mechanisms are involved in the stimulation of cerebral blood flow and endothelial function is important for CBF which should be impaired by blockade of the M3-receptor that release NO from endothelial cells [54]. Brain fog is a very typical complaint in ME/CFS [107]. Potential effects of bradykinin on the blood brain barrier (BBB) should also be considered contributing to CNS symptoms as by increasing microvavscular permeability bradykinin is able to open the blood brain barrier (BBB) and to potentially raise intracranial pressure [66,67].
A frequent complaint is that attention cannot be maintained for a longer time [108]. CBF has been found decreased in ME/CFS patients [109]. Higher CBF and heart rate variability correlated with less severe symptoms [110]. A decrease in CBF may not only impair cognitive function but also affect the motorcortex. This could result in a slowing of movements and uncoordinated movements, which are symptoms of ME/CFS, and fatigue. Significant inverse correlation was seen between CBF and skeletal muscle pH at rest suggesting that mental fatigue and muscular fatigue are related [111].
Paresthesia and burning: A subset of patients suffers from paresthesia and burning mostly in feet and legs. A recent study shows that 40% of 160 patients with ME/CFS with preload failure had evidence of small-fiber polyneuropathy on skin biopsy which has also been found in FM [112]. To explain radicular pain in fibromyalgia it was speculated that a higher cerebrospinal fluid pressure irritates nerve fibres inside nerve root sheaths [113,114]. An increase in BBB-permeability by bradykinin in ME/CFS could indeed cause an excessive production of cerebrospinal fluid and also a higher pressure in the Remak-bundles of unmyelinated fibres (internal compression) potentially affecting perfusion while bradykinin and PGE2 stimulating exactly this type of nerve fibres may simultaneously increase energy consumption. This could result in an energetic deficit and chronic damage. Further there is evidence that autoantibodies can cause small fiber neuropathy which is found in several autoimmune diseases [115].
Sleep disturbances in ME/CFS: Unrefreshing sleep is a typical symptom of ME/CFS. Although patients feel tired they are often unable to fall asleep. PGE2 has awaking effects [75]. PGE2 release is stimulated by bradykinin and ischemic muscles also release prostaglandins [71]. The influence of a potential opening of the BBB by bradykinin on sleep should be considered. A new finding is a high prevalence of obstructive sleep apnea (OSA) in ME/CFS although ME/CFS mainly affects young women who are usually not concerned by OSA [116]. Upper airway mucosal swelling resulting from microvascular leakage increases upper airway resistance increasing the collapse inducing forces to put a higher burden on the upper airway dilating skeletal muscles mainly innervated by the hypoglossal nerve. Those muscles may be subject to fatigue and myalgia as the other muscles in the body. Feeling of a sore throat in in the morning in the absence of pharyngeal lesions (“sterile pharyngitis”) could be explained by myalgic upper airway dilating muscles. Huge sympathetic surges occur in the postapneic phase in OSA. In ME/CFS patients postapneic vasoconstriction may even be stronger than in OSA patients with no ME/CFS due to vascular dysfunction. OSA may strongly aggravate hypoxia in the brain, muscles and other organs which could contribute to fatigue and cognitive dysfunction
4.3. Orthostatic dysfunction (OD), orthostatic stress and the postural tachycardia syndrome (POTS)
Typical ME/CFS symptoms can be provoked not only by increased physical or cognitive activity, but also by orthostatic stress [6]. ME/CFS is associated with POTS which manifests with symptoms of cerebral hypoperfusion and excessive sympathoexcitation [81,117,118]. Orthostatic dysfunction could be due to hypovolemia and preload failure via the algesic vasodilators released from hypoxic muscles after spillover into the arterial system causing renal hyperexcretion and microvascular leakage. Fig. 1 shows the vicious cycle that could arise from these disturbances. The hemodynamic situation when standing up will strongly activate the sympathetic system to cause “orthostatic stress”. Orthostatic intolerance (OI) may bind the patients to beds and wheel chairs. Upright posture consistently aggravates ME/CFS symptoms even in the absence of heart rate and blood pressure changes, often for days, consistent with orthostatic stress being a cause of PEM [6]. The increased sympathetic tone while standing can cause muscular and cerebral vasoconstriction to initiate the cascade of events outlined above for other stressors. The synergistic effect of the simultaneous activation of the baroreflex and the MAA in the early phase of exercise on vasodilator/vasoconstrictor tone in skeletal muscle has been highlighted in the paragraph above.
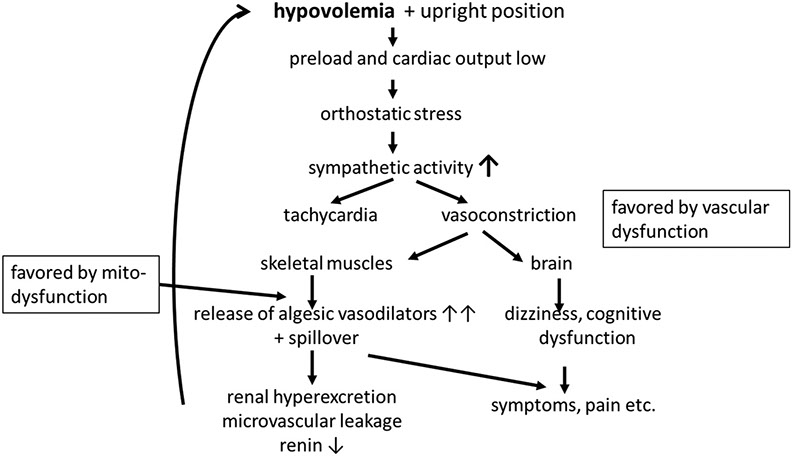
Fig. 1. Presumed pathomechanisms of orthostatic intolerance in ME/CFS.
Due to the suspected dysfunction of ß2AdR heart rate may not even rise proportionally to sympathetic activity (“chronotropic incompetence”) so that the true extent of sympathetic activation and alpha1-adrenergic vasoconstriction may be underestimated by taking the rise in heart rate as a measure for sympathetic activation. An interaction of hyperventilation induced hypocapnia with sympathetically induced paradoxical cerebral vasoconstriction has been reported in a study on the postural tachycardia syndrome (POTS) [119]. In POTS, vasoconstriction is most likely so effective that hypotension does not occur any more. However, at this high level of vasoconstriction cerebral vessels (and vessels of skeletal muscles) may excessively constrict, perhaps favored by hypocapnia, to cause the typical symptoms of POTS or to aggravate the symptoms of ME/CFS. Concerning the frequent association of POTS and ME/CFS causality could be bidirectional. Primary severe orthostatic dysfunction could favor ME/CFS by orthostatic stress as outlined above and, ME/CFS could aggravate orthostatic dysfunction by causing preload failure.
5. Other possible mechanisms of vascular (endothelial) dysfunction in ME/CFS
5.1. Polymorphisms of ß2AdR genes
A mutation (Gln27 mutation) was found associated with (adolescent) chronic fatigue syndrome and a lower HRV was reported [26]. In healthy adults, the Gln27 and the Arg16 mutations of this receptor are associated with blunted regional limb vasodilation, reduced regional and systemic vasodilation to sympathoexcitation, decreased ventricular ejection fraction, cardiac output and stroke volume in line with what has been explained for possible effects of autoantibodies against ß2AdR throughout this manuscript [27]. In an orthostatic challenge the Arg16 variant was found associated with stronger increases in heart rate, systemic vascular resistance and norepinephrine levels and with a decrease in cardiac index [28]. This mutant also showed stronger renal sodium excretion after sodium loading having a potential impact on preload [29]. This pattern of changes has the potential to favor the development of orthostatic dysfunction and POTS. Is it already a mild or asymptomatic form of the CV disturbances seen in ME/CFS?
5.2. The potential role of a high sympathetic tone and vascular (endothelial) dysfunction in ME/CFS
According to heart rate variability studies cited above sympathetic tone is chronically increased in ME/CFS. This could contribute to pathophysiology of ME/CFS by acute and chronic effects. Acute effects include a high vasoconstrictor sympathetic tone which in the presence of endothelial dysfunction and dysfunctional vasodilator ß2AdR could entail exaggerated vasoconstriction that is opposed by the metabolically stimulated release of algesic vasodilators for compensation leading to the symptoms outlined above.
ME/CFS patients show signs of endothelial dysfunction [72,73]. MIAT, which has been found upregulated in PBMC of ME/CFS patients could be an indicator for endothelial dysfunction as elevated MIAT levels in cardiovascular diseases are associated with endothelial dysfunction [120]. Upregulated MIAT in endothelial cells in diabetes is considered a prognostic marker for cardiac remodeling [121]. Endothelial dysfunction might be an important mechanism in ME/CFS by which the vasodilator component of the (enhanced) sympathetic activity may be impaired allowing vasoconstrictor mechanisms mainly carried by alpha1-adrenergic activation to prevail in skeletal muscle and brain. A possible cause are autoantibodies against vasodilator receptors such as ß2AdR and M3-muscarinergic receptors [122,123]. We think that a chronically elevated sympathetic tone itself can lead to endothelial and vascular dysfunction favoring vasoconstrictor influences. Chronic stress induces endothelial dysfunction, indeed [124,125]. Continuous alpha1-adrenergic receptor stimulation causes sensitization of vasoconstrictor mechanisms to calcium and induces growth of VSCMs and adventitial fibroblasts so that the possibility of vascular remodeling should be considered (media hypertrophy) which may enhance the vasoconstrictor effect of the high sympathetic tone in skeletal muscles [126].
A chronically elevated sympathetic tone may also lead to a differential desensitization of adrenergic receptor subtypes with the overall effect that vasoconstrictor influences are increased. Desensitization of β-adrenergic receptors occurs in heart failure due to chronically elevated catecholamine levels [44]. The β2AdR, which is most relevant for muscle and brain perfusion, shows the strongest endocytosis rate via β-arrestin among the different adrenergic receptors causing desensitization [45]. By contrast, the β3-adrenergic receptor and the alpha1-adrenergic receptor, which is the physiologically more relevant vasoconstrictor alpha-adrenergic subtype, do not or only weakly interact with β-arrestin. Altogether, vasodilator ß-adrenergic receptors show stronger desensitization than vasoconstrictor alpha-adrenergic receptors. Desensitization of the ß2AdR may be the most relevant change to ME/CFS. Altogether α1-adrenergic vasoconstriction favored by chronic high sympathetic tone may lead to chronification of ME/CFS. Table 6 gives an overview on possible causes of vascular dysfunction.
Table 6. Possible causes or mechanisms of vascular (endothelial) dysfunction in ME/CFS.

Predisposing and perpetuating factor
Finally, an increased vasoconstrictor tone from different mechanisms would increase the need for functional sympatholysis enhancing the metabolically stimulated generation of algesic vasodilator that after spillover into the systemic circulation cause hypovolemia and the myriad of symptoms of ME/CFS.
5.3. The high prevalence of ME/CFS in patients with craniocervical and atlantoaxial instability
Prevalence of ME/CFS is high in in patients with craniocervical and atlantoaxial instability. When we retrospectively analyzed the history of our patients craniocervical trauma or instability was reported in 24% of a cohort of 133 ME/CFS patients. Hyperventilation occurs after whiplash injury [127]. Hyperventilation in the absence of excitation, hypoxia or hypercapnia could be induced by inappropriate stimulation of chemoreceptors located in the carotid bodies. These act as the main oxygen sensors in the body to mediate the hypoxic respiratory drive. Their stimulation not only increases respiratory drive but also raises sympathetic activity [128]. Since the carotid bodies are in close proximity to the hypermobile cervical spine column it is reasonable to consider the possibility that carotid bodies could be inappropriately activated to stimulate respiration and to raise sympathetic tone (stress). Although the mechanisms of such stimulation of the carotid body chemoreceptors in craniocervical instability are totally unclear our first speculation would be that the (separate) blood vessel system supplying the carotid bodies could be affected - perhaps by distension or compression by the hypermobile cervical spine column - leading to local hypoxia in the carotid bodies despite normal systemic pO2. There may be other causes for chemoreceptor hyperactivity or hypersensitivity than craniocervical instability. For instance, patients suffering from POTS had an increased peripheral chemoreflex in response to hypoxia, which is usually associated with an increase in sympathetic activity, but a decreased central sensitivity to CO2 [129]. Patients frequently report shortness of breath despite normal pulmonary function [130]. Inappropriate stimulation of peripheral chemoreceptors would induce hyperventilation and cause the sensation of dyspnea despite normoxia and normal pulmonary function. Hyperventilation would soon lower pCO2 below the CO2-apneic threshold to induce central apneas which would finally lead to systemic (true) hypoxia towards the end of the apnea raising sympathetic tone again. Inappropriate stimulation of peripheral chemoreceptors during sleep destabilizes breathing to favor central and obstructive apneas which may be another cause for the high prevalence of sleep apnea in ME/CFS patients as outlined above.
Above it has already been highlighted that hypocapnia, which would be induced by inappropriate chemoreceptor stimulation, has vasoconstrictor effects on the cerebral vasculature to lower cerebral blood flow, a mechanism potentially involved in mental fatigue, brain fog and POTS in ME/CFS, which supports sympathetically induced vasoconstriction. We would like to call the increase in sympathetic activity that could arise from inappropriate carotid body stimulation “chemoreceptor stress”. It is a potentially new stressor in patients with ME/CFS that has to be added to the existing list of stressors which already includes physical, emotional, cognitive and orthostatic stress.
6. Conclusion
ß2AdR dysfunction may be an important risk factor in the pathophysiology of ME/CFS potentially caused by dysfunctional autoantibodies to ß2AdR, polymorphisms and desensitization of ß2AdR by (chronic) high sympathetic tone. High sympathetic tone in the presence of endothelial or vascular dysfunction, to which dysfunctional ß2AdR contribute, may lead to an excessive vasoconstrictor stimulus in skeletal muscles, which is opposed by the metabolically stimulated generation of endogenous algesic vasodilators in the process of functional sympatholysis. Metabolic stimulation of the generation of algesic vasodilators is strongly favored by the poor background energetic situation in skeletal muscle which is due to a still enigmatic metabolic disturbance (probably a mitochondrial disturbance). Spillover of such excessively generated algesic vasodilators into the circulation could explain the unfavorable CV situation causing hypovolemia, inhibition of renin production and preload failure further raising sympathetic (vasoconstrictor) tone. The appearance of algesic vasodilators in the systemic circulation may explain many of the enigmatic symptoms of ME/CFS including pain, hyperalgesia, intestinal complaints, flu-like symptoms, sore lymph nodes and others.
Close window by clicking in grey area >