Stem cell application in neurorehabilitation
Over the last 35 years major advances have been made in the field of neural restoration and this includes not only better strategies for endogenous repair but also the ability to actively intervene through neural grafting for example (see Table 14.1). This revolution stems from a better understanding of the processes underlying intrinsic repair, the realization that endogenous processes such as neurogenesis still occur in the adult mammalian brain [1] , and that neurotrophic factors can be used to encourage cell survival and fibre outgrowth in diseased cells within the central nervous system (CNS), along with in situ reprogramming (reviewed in [2]). In addition, we have realized that transplant of certain cells can survive in the adult CNS and make and receive connections with functional benefits to the grafted animal. In this chapter we explore various aspects of these processes including the role of adult neurogenesis in health and disease as well as the cell based approaches that have been used to treat a whole variety of CNS disorders but especially Parkinson’s and Huntington’s disease and spinal cord injury (see Box 14.1, and [2–4]).
Basic biology of stem cells
Endogenous neurogenesis in the adult mammalian brain It has been a long held concept in the neurosciences that the generation of neurons tapers off with the end of embryonic development. For decades the leading opinion in the field was that the adult mammalian brain is not capable of generating new neurons throughout life due to the absence of any neurogenic, dividing cells, i.e. neural stem cells (NSCs). It was assumed that the neuronal networks and circuitries in the mature CNS are too complex to allow for the maturation and integration of newborn neuronal cells. Thus, the most common strategy to ameliorate disease symptoms and promote rehabilitation in the context of neuropsychiatric disease was to enhance functional plasticity of surviving networks and brain areas. However, the concept that the adult brain loses its capacity to regenerate was challenged in the mid 1960ies when the first experiments suggested that there may be proliferating cells in the adult brain that appeared to have the potential to generate new neurons, at least in distinct areas of the mammalian brain [5, 6]. In these pioneering studies by Altman, Kaplan, and others radioactively labelled thymidine was used to visualize proliferating cells doubling their DNA content prior to cytokinesis. However at this time it was technically extremely difficult to truly confirm that i) a cell is newborn and
ii) differentiates into a neuron. However, the idea that the adult brain may even be capable of generating new neurons was fueled by the findings that cells could be isolated and propagated in vitro that showed NSC properties meaning that these cells were able to self renew and to generate neurons in the culture dish [7] .
This technical breakthrough, leading to the acceptance that neurogenesis occurs in the mammalian brain throughout life, came with the use of thymidine analogues (such as bromodeoxyuridine; BrdU) that could be visualized using antibodies in combination with techniques to label neuronal cells using confocal microscopy (e.g. [8]).
With this strategy—that was later complemented using specific retroviruses that selectively label dividing cells and their progeny and transgenesis based approaches—two main neurogenic regions in the adult mammalian brain could be identified: the subventricular zone (SVZ) lining the lateral ventricles out of which newborn cells migrate along the rostral migratory stream (RMS) towards the olfactory bulb (OB) where they differentiate into several types of mostly GABAergic olfactory neurons, and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) where exclusively glutamatergic, excitatory granule cells are generated [9] . In these two neurogenic areas NSCs with certain astrocytic properties that are largely quiescent (i.e. do rarely divide) under normal conditions give rise to more proliferative progenitors that generate new, immature neurons [10, 11]. These new neurons structurally and functionally mature over the course of several weeks before they integrate into the preexisting neural circuitries in the OB and DG. Interestingly, the functional properties of young, immature neurons substantially differ from their older progeny that are generated during embryonic development, as newborn cells are much more excitable and display a higher degree of plasticity which is believed to be the reason why the adult brain invests in the energetically demanding exercise of supporting these two neurogenic (and highly plastic) regions [10].
Notably, the number of neurons generated is not static but rather is dynamically regulated. Positive stimuli such as physical exercise and environmental enrichment strongly enhance the number of newborn neurons whereas negative regulators such as stress and ageing substantially decrease neurogenesis [10]. Initially based on these correlative data it was hypothesized that adult neurogenesis may be not only important for physiological brain function but may also contribute to certain disease processes—for example in the context of affective disorders such as major depression as well as neurodegenerative disorders [12]. Indeed, in mouse models of stress and depression it could be shown that certain antidepressants strongly enhanced neurogenesis and their behavioural effect is dependent on pharmacologically enhanced neurogenesis [13]. Besides a contribution of altered or failing neurogenesis to certain disease processes the identification of endogenous NSCs also opened up the possibility of activating and recruiting NSCs or their neuronal progeny to lesioned or injured brain areas to replace lost neurons (e.g. in the context of ischaemic stroke). Thus, targeting endogenous NSCs that generate new neurons throughout life presented a novel treatment option to improve or restore brain function in a number of neuropsychiatric diseases.
Exogenous stem cells for neural repair
There have been numerous attempts both experimentally as well as clinically over the last decades to replace lost neurons not only by mobilizing endogenous NSCs but also by transplanting exogenous cells with the capacity to produce new neurons into the diseased or injured brain. For example, fetal progenitors were used to replace lost dopaminergic neurons in the context of Parkinson’s disease (PD) (for details, see Stem cell based therapeutic approaches in Parkinson’s disease, p. 172). However, it soon became evident that the heterogeneity of transplants and the inherent difficulties in standardizing the dissection and preparation of the tissue as well as ethical issues made it extremely challenging to use fetal human progenitors as a standard treatment option in PD. Thus, new cellular sources had to be identified and developed that could restore and replace specific neural structures lost to brain injury/degeneration.
Much hope to find a reliable source for neuronal cell replacement was invested in human embryonic stem cells (ESCs). ESCs are derived from the inner cell mass early during embryonic development, and represent a cell type that shows pluripotency—which means that these cells are capable of generating all tissues of the organism—(besides the trophoblast)—along with the capacity for almost indefinite self renewal. In the last decade, substantial progress has been made to develop protocols to direct ESCs towards specific neuronal lineages, such as dopaminergic neurons (e.g. [14, 15]).
These protocols have also brought with them reassurance that the risk of transplanting undifferentiated, and thus dividing, ESCs with the potential to form teratomas has now been circumvented. Nevertheless, the clinical use of ESCs is still challenged by ethical concerns (given that human ESCs are derived from the progeny of vitro fertilized oocytes) and the fact that transplanted ESCs are nonautologous transplants requiring at least a certain degree of immunosuppression to prevent rejection of the transplanted tissue by the host.
Given these limitations, the discovery that virtually every somatic cell can be reprogrammed to adopt a pluripotent state by introducing defined transcription factors opened novel possibilities for patientspecific cell replacement strategies. These cells, called induced pluri-potent stem cells (iPSCs), can be easily generated from each individual (e.g. by a simple skin biopsy and isolating fibroblasts that are then subjected to reprogramming) yielding an isogenic and patient-selective source for therapeutic cell replacement strategies [16]. However, the use of these cells in an autologous fashion for patients is still some way off, given the cost of their manufacture and the cost that comes with the preclinical testing of any one stem cell line product.
In the following sections we will briefly review the evidence that altered neurogenesis in the adult brain contributes to neuropsychiatric disease processes and how the mobilization of endogenous or the use of exogenous, transplanted stem cells may provide novel treatment options in the context of neurodegeneration, specifically in Parkinson’s and Huntington’s disease, as well as other CNS dis-orders such as spinal cord injury
Basic biology of stem cells
Endogenous neurogenesis in the adult mammalian brain It has been a long held concept in the neurosciences that the generation of neurons tapers off with the end of embryonic development. For decades the leading opinion in the field was that the adult mammalian brain is not capable of generating new neurons throughout life due to the absence of any neurogenic, dividing cells, i.e. neural stem cells (NSCs). It was assumed that the neuronal networks and circuitries in the mature CNS are too complex to allow for the maturation and integration of newborn neuronal cells. Thus, the most common strategy to ameliorate disease symptoms and promote rehabilitation in the context of neuropsychiatric disease was to enhance functional plasticity of surviving networks and brain areas. However, the concept that the adult brain loses its capacity to regenerate was challenged in the mid 1960ies when the first experiments suggested that there may be proliferating cells in the adult brain that appeared to have the potential to generate new neurons, at least in distinct areas of the mammalian brain [5, 6]. In these pioneering studies by Altman, Kaplan, and others radioactively labelled thymidine was used to visualize proliferating cells doubling their DNA content prior to cytokinesis. However at this time it was technically extremely difficult to truly confirm that i) a cell is newborn and
ii) differentiates into a neuron. However, the idea that the adult brain may even be capable of generating new neurons was fueled by the findings that cells could be isolated and propagated in vitro that showed NSC properties meaning that these cells were able to self renew and to generate neurons in the culture dish [7
With this strategy—that was later complemented using specific retroviruses that selectively label dividing cells and their progeny and transgenesis based approaches—two main neurogenic regions in the adult mammalian brain could be identified: the subventricular zone (SVZ) lining the lateral ventricles out of which newborn cells migrate along the rostral migratory stream (RMS) towards the olfactory bulb (OB) where they differentiate into several types of mostly GABAergic olfactory neurons, and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) where exclusively glutamatergic, excitatory granule cells are generated [9] . In these two neurogenic areas NSCs with certain astrocytic properties that are largely quiescent (i.e. do rarely divide) under normal conditions give rise to more proliferative progenitors that generate new, immature neurons [10, 11]. These new neurons structurally and functionally mature over the course of several weeks before they integrate into the preexisting neural circuitries in the OB and DG. Interestingly, the functional properties of young, immature neurons substantially differ from their older progeny that are generated during embryonic development, as newborn cells are much more excitable and display a higher degree of plasticity which is believed to be the reason why the adult brain invests in the energetically demanding exercise of supporting these two neurogenic (and highly plastic) regions [10

Notably, the number of neurons generated is not static but rather is dynamically regulated. Positive stimuli such as physical exercise and environmental enrichment strongly enhance the number of newborn neurons whereas negative regulators such as stress and ageing substantially decrease neurogenesis [10]. Initially based on these correlative data it was hypothesized that adult neurogenesis may be not only important for physiological brain function but may also contribute to certain disease processes—for example in the context of affective disorders such as major depression as well as neurodegenerative disorders [12]. Indeed, in mouse models of stress and depression it could be shown that certain antidepressants strongly enhanced neurogenesis and their behavioural effect is dependent on pharmacologically enhanced neurogenesis [13]. Besides a contribution of altered or failing neurogenesis to certain disease processes the identification of endogenous NSCs also opened up the possibility of activating and recruiting NSCs or their neuronal progeny to lesioned or injured brain areas to replace lost neurons (e.g. in the context of ischaemic stroke). Thus, targeting endogenous NSCs that generate new neurons throughout life presented a novel treatment option to improve or restore brain function in a number of neuropsychiatric diseases

Exogenous stem cells for neural repair
There have been numerous attempts both experimentally as well as clinically over the last decades to replace lost neurons not only by mobilizing endogenous NSCs but also by transplanting exogenous cells with the capacity to produce new neurons into the diseased or injured brain. For example, fetal progenitors were used to replace lost dopaminergic neurons in the context of Parkinson’s disease (PD) (for details, see Stem cell based therapeutic approaches in Parkinson’s disease, p. 172). However, it soon became evident that the heterogeneity of transplants and the inherent difficulties in standardizing the dissection and preparation of the tissue as well as ethical issues made it extremely challenging to use fetal human progenitors as a standard treatment option in PD. Thus, new cellular sources had to be identified and developed that could restore and replace specific neural structures lost to brain injury/degeneration.
Much hope to find a reliable source for neuronal cell replacement was invested in human embryonic stem cells (ESCs). ESCs are derived from the inner cell mass early during embryonic development, and represent a cell type that shows pluripotency—which means that these cells are capable of generating all tissues of the organism—(besides the trophoblast)—along with the capacity for almost indefinite self renewal. In the last decade, substantial progress has been made to develop protocols to direct ESCs towards specific neuronal lineages, such as dopaminergic neurons (e.g. [14, 15]).
These protocols have also brought with them reassurance that the risk of transplanting undifferentiated, and thus dividing, ESCs with the potential to form teratomas has now been circumvented. Nevertheless, the clinical use of ESCs is still challenged by ethical concerns (given that human ESCs are derived from the progeny of vitro fertilized oocytes) and the fact that transplanted ESCs are nonautologous transplants requiring at least a certain degree of immunosuppression to prevent rejection of the transplanted tissue by the host.
Given these limitations, the discovery that virtually every somatic cell can be reprogrammed to adopt a pluripotent state by introducing defined transcription factors opened novel possibilities for patientspecific cell replacement strategies. These cells, called induced pluri-potent stem cells (iPSCs), can be easily generated from each individual (e.g. by a simple skin biopsy and isolating fibroblasts that are then subjected to reprogramming) yielding an isogenic and patient-selective source for therapeutic cell replacement strategies [16]. However, the use of these cells in an autologous fashion for patients is still some way off, given the cost of their manufacture and the cost that comes with the preclinical testing of any one stem cell line product.
In the following sections we will briefly review the evidence that altered neurogenesis in the adult brain contributes to neuropsychiatric disease processes and how the mobilization of endogenous or the use of exogenous, transplanted stem cells may provide novel treatment options in the context of neurodegeneration, specifically in Parkinson’s and Huntington’s disease, as well as other CNS dis-orders such as spinal cord injury
Therapeutic targeting of neural stem cells in neuropsychiatric disease
Adult hippocampal neurogenesis and affective disorders Affective disorders such as major depression generate a major social and financial burden on Western societies given their high prevalence. Even though several classes of antidepressant drugs have been in clinical use for decades, there remains a substantial fraction of patients with therapy resistant disease, indicating the need to
i) better understand the aetiology and neural consequences of affective disorders and
ii) novel treatment strategies. One of the key risk factors (besides age, see Age associated cognitive decline and reduced neurogenesis, p. 171) to develop affective disorders is stress, which was found to dramatically reduce neurogenesis [18]. In combination with the findings that a structural hallmark of patients suffering from depression is the reduction of hippocampal volume as measured by noninvasive imaging approaches, this initiated a large number of studies aiming at understanding a potential link between the onset or maintenance of affective disorders and adult hippocampal neurogenesis [13, 19, 20]. Strikingly, a number of antidepressants such as selective serotonin reuptake inhibitors (SSRIs) enhance the number of neurons generated and depend at least partially on neurogenesis for their efficacy (e.g. [21]). Supporting the potential contribution of failing neurogenesis to the depressive disease process has been the fact that many antidepressants show a latency of 2–4 weeks between being taken and having a therapeutic effect—a time that may reflect the antidepressant induced generation, maturation, and functional integration of newborn neurons [13].
However, genetic enhancement of hippocampal neurogenesis turned in the adult hippocampus represents a novel therapeutic target to ameliorate disease symptoms in major depressive disorders. On the other hand, it is unlikely that hippocampal neurogenesis is the major and sole cause whose alterations may lead to or, if pharmacologically targeted, cure depression. Most importantly, more evidence needs to be produced that neurogenesis may indeed be affected in patients suffering from affective disorders.
Age associated cognitive decline and reduced neurogenesis Ageing is associated with a substantial decline in several cognitive domains that may eventually lead to severe disabilities [23, 24].
Interestingly, the number of neurons that are generated in the adult hippocampus (and SVZ/OB system) dramatically decreases with advancing age (without coming to a complete stop) [1, 10], even though these findings remain controversial [25–27]. Furthermore, the number of neurons born in older age does correlate with the performance of rodents on hippocampus dependent learning tasks [28]. Thus, it has been speculated that hippocampal neurogenesis may be a critical mediator of cognition with ageing. This idea has been supported for example by imaging based findings in humans that the first structure showing functional and structural alterations in cognitively challenged, aged individuals is indeed the hippocampal DG [29]. In addition, known regulators of neurogenesis such as running and environmental enrichment have turned out to be effective in enhancing neurogenesis in aged rodents, which was again associated with improved performance in hippocampus dependent learning tasks [30, 31
Current projects aim to elucidate the cellular and molecular mechanisms that are responsible for the age dependent drop of neurogenesis. Furthermore, it remains unclear if enhancing neurogenesis is sufficient to ameliorate cognition in advanced age. Be that as it may, the observed association between cognitive decline and decreased neurogenesis suggests a mechanism at least partially explaining the drop in hippocampus dependent cognition with old age.
Altered neurogenesis in epilepsy
Besides the abovementioned diseases that reduce the number of neurons, there are also disease states that at least transiently enhance the number of neurons generated. For example, it has been shown that neurogenesis is dramatically enhanced in rodent models of temporal lobe epilepsy (TLE) [32]. Notably, not only is the number of neurons generated enhanced: but epileptic activity also leads to ectopic migration of newborn granule cells into the hilar region of the DG and the aberrant formation of hilar basal dendrites that form ectopic synapses and potentially impair proper synaptic transmission and connectivity within the dentate circuitry [33, 34]. Strikingly, it has also been shown that ectopic neurogenesis is sufficient to drive epilepto genesis, further supporting the findings that enhanced but massively altered neurogenesis in the context of TLE is potentially a contributing disease factor [35]. However, neurogenesis may not only be involved in the establishment of epileptogenic circuitries as it has also been shown that in more advanced or chronic disease stages, neurogenesis is strongly downregulated and may represent one factor responsible for cognitive decline that is commonly observed in patients affected by chronic or therapy refractory forms of TLE [36]. Thus, drugs aiming to enhance neurogenesis in the context of affective disorders or ageing may also turn out to be effective in ameliorating cognitive symptoms in advanced stages of TLE by increasing the number of newborn granule cells [37
This has been looked at in animal models of TLE through the transplantation of exogenous NSCs into the epileptic hippocampus. First results are promising even though the invasiveness and associated risks when considering the next steps in taking such a strategy into the clinical setting are substantial [38
Stem cell based therapeutic approaches in Parkinson’s disease Parkinson’s disease is a common neurodegenerative disorder of the CNS that affects about 1 in 800 people and typically presents in late life around 70 years of age. It has as part of its core pathology the loss of the nigrostriatal dopaminergic neurons and the formation of alpha synuclein positive Lewy bodies. However, in recent years a number of fundamental new concepts have emerged with respect to PD:
1. The disease process is not restricted to dopaminergic nigrostriatal neurons but involves many sites within the brain and even neurons outside of the CNS (e.g. in the enteric nervous system) [39];
2. PD is not simply a disorder affecting motor control but embraces a range of nonmotor features, some of which may even precede the onset of the movement disorder (so called prodromal or premotor PD) [40–42]);
3. The disease process may even begin in the periphery and then spread into the CNS with alpha synuclein behaving in a prion like fashion [43–45];
4. While the pathogenesis of the disease process may involve protein spread, it is still unknown why people develop PD in the first place although there is now substantial evidence to show that there are major genetic risk factors for getting it [46]. This includes heterozygote mutations in genes coding for glucocerebrosidase (GBA), in which homozygous mutations cause Gaucher’s disease [47, 48];
5. There are now a large number of mendelian forms of PD described some of which resemble idiopathic PD both clinically and pathologically [49];
6. Idiopathic PD is heterogeneous and the basis for this may relate to genetic variants that are also linked to the risk of getting it in the first place [50, 51
All of this has had implications for the use of stem cells in the study and treatment of PD in two main ways:
i. Disease modelling using iPSCs, typically from patients with Mendelian forms of PD or stem cell lines transfected by the gene of interest [52];
ii. The fact that neural grafting with dopaminergic cell transplants will only help some patients with PD and then only some of their symptoms and signs—in other words, it will never be a treatment for all patients with PD [2, 53].
PD disease modelling
iPSCs derived from patients offer a powerful in vitro disease model as these should carry the identical cellular pathological features of the disease of that patient [54]. However, there are several key assumptions with this approach. Firstly, that any pathology seen in neurons so derived after a few days or weeks in culture is disease relevant and speaks to the pathology seen in the CNS of the patient that has taken decades to develop. Secondly it assumes that the disease process is cell autonomous as the only cells being studied are the neurons so generated, and it does not, and cannot, interrogate how different cellular players may talk to each other in the disease process (e.g. the role of inflammation). Thirdly the reprogramming of the cells removes some of the age related factors [55] that are critical in the development of PD, given this is the biggest risk factor for PD. Finally, it has to be shown that the neurons so produced are truly authentic neurons of the type wanted. This is especially true for dopaminergic neurons as there are at least 10 subtypes of dopaminergic neurons in the adult brain (A8– A17) [56], all of which show specific electrophysiological, neurochemical and transcriptional profiles and only some of which are lost in PD [57
Despite a number of obstacles, several groups have produced PD patient specific iDA derived from iPSCs and early studies found that the differentiated cells did not show any disease related phenotype [58]. In addition, it was noted that residual transgene expression in virus carrying iPSCs influenced their molecular properties, which has led to the use of derivation methods free of reprogramming factors in modelling of human diseases. Subsequently it has been shown that iDA derived from iPSCs do display specific PD pathology using cell lines from patients with sporadic and LRRK2associated PD [59, 60]. As with the earlier study [58], no difference was observed between the iDA from PD patients and controls in the differentiation efficiency, morphology, and phenotype after 30 days in culture. However, longterm culture (<75 days) of iDA derived from sporadic PD cases revealed altered morphology, with a decrease in the number and length of neurites and an increased susceptibility to degeneration. iDA from PD patients also exhibit defective autophagosome clearance [59], as well as mitochondrial defects [61, 62
While the vast majority of PD cases are idiopathic (>95%), several causative genes have been identified in families harbouring mendelian forms of the disease [63]. So far at least five PD related genes have been studied using iPS cell technology including neurons derived from patients carrying mutations in the alpha synuclein (SNCA), glucocerebrosidase, Leucine Rich Repeat Kinase2 (LRRK2), phosphatase, and tensin homologue (PTEN)induced putative kinase 1 (PINK1) and Parkin genes. All of these have shown some pathological changes, although they are often subtle and their relevance to the disease process in the affected patient is unclear
In recent years, neurons differentiated from iPSCs while providing new insights into the cellular mechanisms involved in the pathophysiology of PD, have also been considered for transplantation. However, concerns remain with respect to their safety, mainly due to their proliferative, tumorigenic potential [64, 65], but of late these anxieties have been allayed by longterm studies in nonhuman primates using dopaminergic neurons made from iPSCs derived from control subjects and PD patient [66]. To overcome this issue, several groups have developed methods that allow direct conversion of human differentiated somatic cells, such as fibroblasts, into functional neurons avoiding any intermediate pluripotent state. The first study to do this converted mouse embryonic and postnatal fibroblasts into functional neurons by the overexpression of three transcription factors (Ascl1, Brn2, and Mytl1) [67]. Subsequently human fibroblasts have also been successfully converted into functional neurons by overexpressing the same transcription factors [68] and this has now also been done in disease conditions (e.g. Alzheimer’s disease patients) [69]. For PD, obviously making dopaminergic neurons would be of interest and it has been shown that the addition of two transcription factors specific to dopaminergic lineage (Lmx1a and FoxA2) to the three original factors is sufficient to generate dopaminergic like (DA) neurons [68, 70]. However, the gene expression profiles of these reprogrammed DA neurons differed significantly from primary midbrain DA neurons in these studies and so more recent attempts to generate iDA like midbrain dopaminergic neurons have used six reprogramming factors (Ascl1, Pitx3, Nurr1, Lmx1a, Foxa2, and En1), as well as the patterning factors Shh and FGF8 [71]. While these iDA expressed many of the relevant markers of dopaminergic neurons, the cells only partially restored dopamine function in vivo, and have failed to exhibit similar levels of midbrain transcription levels found in embryonic or adult midbrain dopamine neurons [71]. More recently, a combination of five transcription factors (Ascl1, Pitx3, Nurr1, Sox2, and Ngn2) generated iDA that further provided benefit when grafted in the 6OHDA rat model of PD, suggesting that these reprogrammed cells display functional midbrain dopaminergic neuronal properties [72
Because the direct conversion does not go through a proliferative state, the quantity of neurons that can be obtained is limited by the accessible number of fibroblasts used as starting material for conversion. Nevertheless, direct conversion of the patient’s fibroblasts into relevant neuronal subtypes is very promising for disease modelling and may even ultimately have a role in neural grafting [73].
Neural transplantation
The core loss of the dopaminergic neurons in PD coupled to the response of patients to dopaminergic drugs led in the 1980s to the idea that this condition could be treated through the transplantation of dopaminergic cells into the diseased basal ganglia. This initially involved autografts of the catecholamine rich adrenal medulla, although the results were generally disappointing both experimentally and in patients. It was therefore not long before this approach was superseded by transplants of fetal ventral mesencephalic tissue
[74]. This approach involves harvesting the dopaminergic neurons from the developing ventral midbrain and then grafting them into the site where dopamine normally works, namely the striatum.
Experimentally it was shown that this approach worked well when the cells were harvested at the time they normally develop (E13–14 in rats and mice; 6–8 weeks post conception in humans) as the grafted cells could survive, make and receive connections from the host brain and release dopamine in a regulated manner with functional benefits to the grafted animal. It was on this background that open label studies were undertaken in patients with PD both in Europe and the United States. These studies showed that some patients could derive longterm benefit from these grafts and these clinical improvements correlated with Fdopa positron emission tomography (PET) imaging evidence of dopamine cell survival at the site of implantation. A correlation that was confirmed in a few postmortem studies [75]. The success of this approach gave confidence in some quarters to push on and undertake more rigorous doubleblind placebo controlled trials even though the results from the open label studies had been variable and the optimal way of giving the therapy not resolved [76
These two doubleblind placebo control trials that were published in 2001 and 2003 showed that the therapies were ineffective in so much as they failed to deliver on their primary end point [77, 78].
Furthermore, significant numbers of patients developed involuntary movements in the absence of Ldopa but in the presence of the graft; so called graft induced dyskinesias. Thus in many eyes it was shown that this approach did not work, produced side effects and subsequently it was also shown that the transplants even develop the pathology they are designed to treat [79].
However a more critical review of the trial data leads one to a rather different conclusion, which is that the fetal ventral mesencephalic (VM) grafts can work very well in some patients and that it is understanding why that determines whether this whole approach has a future (reviewed in [80]). However the use of human fetal tissue as the source of cells for grafting is clearly not possible in the long term for a range of ethical and practical reasons and as such there is a need to find a more ethically acceptable, readily available source of dopaminergic neurons for grafting [14, 81] (see Table 14.2
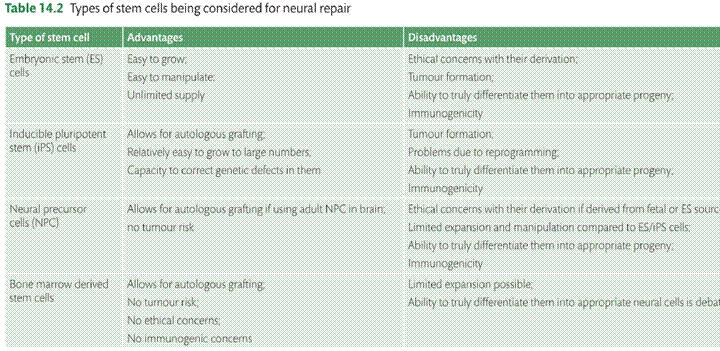
One such cell source are ESCs [82]. However, the use of these cells has been hampered by problems of cell overgrowth; immune rejection and the ability to truly direct them into authentic nigral dopaminergic neurons. Of late though advances have been made in this area with the production of large numbers of A9 looking nigral neurons, which can survive grafting in animal models of PD with functional benefits and no tumour formation [14, 15, 83]. As an alternative iDA generated from iPSCs derived from patients’ skin fibroblasts are very appealing candidates [84], not only because they circumvent ethical issues but exclude the risk of immune rejection. One other benefit in using iPS cells is the possibility of rejuvenating the cells from an aged patient and thus eliminating the pathologies associated with ageing to restore tissue proliferation and function. The potential of iDA derived from iPS cells for cell replacement therapy has been assessed [85], and of late they seem to behave as well as embryonic stem (ES) derived dopamine cell products [66]. More recently it has been shown that differentiated iN and iDA can have effects in the 6OHDA lesioned rat, but the effects are modest at best with the cells not looking like mature nigral neurons [71]. Finally, there have been attempts to circumvent the need for an exogenous cell source for repairing the PD brain, through using approaches that work on in situ reprogramming [86]. However, to date the effects have been rather modest compared to stem cell derived transplants
Stem cell based therapeutic approaches in Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant disorder in which the abnormal gene codes for a mutant huntingtin protein that is expressed in every cell of the body. The disease typically presents in midlife with a combination of motor, cognitive and psychiatric problems and it then progresses over a 20–25 year period to death [87, 88]. The pathological changes become more widespread with disease progression and while it was initially thought that the striatum was the main site of pathology in early HD, this view is in need of qualification based on the results of recent studies in early and premanifest HD [89]. These studies have shown that while the striatum is an early site of pathology, many other areas are affected, which is important given that the transplant approach to HD has only concentrated on repairing the medium spiny output neurons of the striatum. Of interest in this respect is the recent evidence suggesting that striatal neurons seem to be generated throughout life in the human striatum, and that this is reduced in HD patients. This raises the possibility that targeting endogenous neurogenic cells to enhance for example cell genesis and proliferation—in addition to transplantation based strategies—may hold potential for future therapeutic avenues in this condition [90
In the 1980–1990s (before the gene for HD was discovered and thus the advent of HD transgenic mice) it was shown experimentally that grafts of fetal striatal tissue placed in the excitotoxic lesioned striatum could survive, differentiate, receive and make synaptic connections with the host brain and restore behaviour (reviewed in [91]—results which have been less impressive in transgenic animal models of disease [92, 93].
Thus, based on the work in the nontransgenic models of HD, early clinical trials were done using human fetal striatal allografts in patients with mild moderate disease. These trials were most notably done in the United States and Europe and showed mixed results [94].
In the first major study to report, the French group found that 3of their 5 grafted patients showed some transient benefits and that these were linked to evidence of metabolic activity at the site of transplantation [95]. This was followed by a negative outcome study from the group based in South Florida where they found no benefit in any of their patients [96]. This transplant trial used a different approach with respect to the tissue dissection and this could help explain why they did not find any benefits. Interesting of late though, there has been work showing that the grafts in these patients have pathology resembling that seen in the host HD brain [97]. Other studies, most notably one in the United Kingdom, have tended to show that the approach using the current protocols are largely unsuccessful [80], although occasional successes have been seen in individual cases [98]. This variability is now being further explored in a large clinical study in France.
While it is unclear whether fetal striatal allografting is useful and even sensible in HD, it has nevertheless led many to look at making striatal output neurons from stem cell sources for possible use in this way [99]. While to date the number of studies doing this have been limited, it is encouraging that the ability to make these cells is possible and that they do have some benefits in animal models of HD [100–102]. In addition stem cells are also being developed for use in HD through nonneuronal replacement strategies including using neural precursor cells for trophic support [103] and astrocytic grafts to support the HD affected neurons [104].
An alternative use of these cells is to study disease pathogenesis in much the same way as has been done in PD. Thus, work has been done using stem cells transfected with part of the mutant huntingtin gene and more recently neurons derived from iPS cells from HD patients have been produced [105]. This has helped confirm some of the key steps in the disease process, and while these cells have not as yet been thought of as being useful in autografting therapies, this may evolve if the technologies for correcting gene defects can be perfected.
Finally, the ability to use stem cells to repair the brain from within has always held great attraction since it was first shown that adult neurogenesis occurs in the mammalian brain (see earlier). In the case of animal models of HD it has been shown that abnormalities exist in hippocampal neurogenesis and this may account for some of the cognitive and affective aspects of the disease [106–108]. While the basis of this and its relevance to human disease is unknown, it does suggest that intrinsic repair strategies around this system may offer some potential therapeutic avenues worth exploring. In addition there have some reports of increased neurogenesis in the SVZ in HD [109], although again the relevance and significance of this is unknown as are changes in this same system seen in PD and mediated through a midbrain dopaminergic projection and ciliary neurotrophic factor (CNTF) and epidermal growth factor receptor (EGF) signaling pathways (see, for example, O’Keeffe et al. [110]).
Stem cell based therapeutic approaches in stroke
Stroke is a common disorder that affects many people and encompasses a range of different pathologies from large vessel occlusions with hemispheric loss of tissue to small vessel events causing lacunar infarcts. As a result, there has been much interest in using cells to repair the brain in stroke, although exactly how these cells might do this is debatable. Indeed, the idea has been pursued that they could be used for cell replacement, although this seems unlikely to work given the complexity and diversity of cells lost as part of the original insult. Nevertheless, there has been great interest in developing therapies that either recruit endogenous stem cells for repair or the implantation of exogenous grafts of stem cell derived progenitors that work to enhance repair through some form of paracrine effect
After experimental stroke in rodents (e.g. occlusion of the middle cerebral artery; MCAO) proliferation of NSCs in the SVZ is strongly enhanced and a small fraction of these newborn cells can migrate away from the SVZ towards the striatum where they differentiate into neuronal cells (e.g. [111]). Similar observations have been made in human samples [112]. However, at this time it remains unclear if stroke induced endogenous neurogenesis functionally contributes to recovery [113]. Furthermore, it appears that the number of neurons generated is very low. Thus, strategies need to be developed that either enhance the survival or increase the recruitment of newborn cells generated from endogenous NSCs towards the ischaemic lesion.
In the case of neural grafting, this has now evolved to the level of early clinical trials even though their experimental basis is often not that convincing. The most recent of these is a small open label study by ReNeuron in which implants of their immortalized human cortical cell line have been delivered to patients with well established infarcts. The preliminary published data shows that this approach seems safe with some signal of efficacy [114]. This is not the first trial using this approach as various other small open label studies have been undertaken. However, none have produced robust enough effects to be confident that these cells have a future in the treatment of this common condition (see [115
Stem cell based therapeutic approaches in multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease that leads to chronic demyelination followed by axonal loss and a loss of neuronal function. Stem cell based strategies (outside of bone marrow transplantation) to ameliorate disease progression and/or clinical symptoms have been trialed (e.g. [116, 117], reviewed in [118]) and may work through modulation of the autoimmune response and stem cell mediated regeneration [119, 120].
Interestingly, peripheral administration of NSCs and mesenchymal stem cells (MSCs) seems to attenuate the immune reaction in animal models of MS such as experimental autoimmune encephalomyelitis (EAE) most probably by interfering with B cell proliferation and promoting T cell anergy. This leads to fewer inflammatory infiltrates and slowed disease progression in EAE. How stem cells exactly mediate these effects remains largely unknown but given that they can be easily delivered into the periphery this approach could translate into the clinical setting rather rapidly if proven preclinically to be of value [120].
However, a large problem in MS is the degeneration of axons followed by neuronal dysfunction that eventually occurs even in the absence of a strong inflammatory state. Thus, strategies aiming to enhance remyelination are urgently required to truly advance regeneration in MS brains. Again in preclinical EAE models, the transplantation of stem cells with the ability to differentiate into oligodendrocytes reduced disease features. Furthermore, the targeted differentiation into myelinating oligodendrocytes derived from endogenous NSCs in the murine SVZ turned also out to be beneficial in EAE (e.g. [121]). Thus, current experiments aim to identify small molecules that may enhance the endogenous generation of functional oligodendrocytes to ameliorate disease features in chronic demyelinating disease [122, 123].
Stem cells have also been used in other neurological conditions including multiple system atrophy (MSA). In this disease MSCs from the bone marrow have been used and in all cases the benefits seem marginal and need confirming in other studies [124]. The rationale for this approach is that these cells can have immune modulating effects as well as releasing trophic factors, all of which can help repair the brain. The same is also true for the adoption of similar strategies in motor neuron disease [125], and in this respect there is an ongoing trial being led by Clive Svendsen in California looking at glial cell derived neurotrophic factor (GDNF) expressing neural precursor cells implanted adjacent to the lumbar spine in patients with established amyotrophic lateral sclerosis (ALS
Stem cell based approaches in traumatic spinal cord injury
Spinal cord injury (SCI) is a rare disorder (incidence ranges from 15 to 30/million of the population) [126, 127] and in many countries regulatory offices grant an orphan disorder designation to it. Due to an increased level of life expectancy achieved over the last three decades (overall life expectancy depending on the level of lesion is about 90% compared to age matched controls) the prevalence of people living with SCI is steadily increasing (in the United States with an estimated incidence of about 12
000 new cases per year and the prevalence of patients living with SCI is about 1 million of the population) [128, 129]. In about 50% of patients the SCI is due to a traumatic event and for this specific population of patients, rehabilitation standards and outcome assessments have been continuously developed since the first conception and installation of a dedicated SCI rehabilitation programme (in Stoke Mandeville UK, 1942; see also [130, 131]). Traumatic SCI typically affects healthy and younger subjects (although in the recent decades a shift towards elderly subjects is observed with an increase of the mean age from 30 to 45 years), and compared to other neurological disorders of the brain (like MS, stroke, etc.) clinically constitutes a nondegenerative and nonprogressive disorder [132–134]. However, recent neuroimaging studies reveal ongoing supralesional signs of neural degeneration at the level of the (cervical) cord, brainstem, and brain following acute SCI. The impact on clinical recovery and interaction with rehabilitation strategies (physiotherapeutic and drug/cell interventions) remains to be explored [135–139]. Furthermore, SCI represents a very distinct disorder within the CNS due to a rather localized lesion within the cord. Although the spinal cord is embedded in the CNS compartment (sealed by the blood brain barrier) the SCI also clinically affects important neural tissue belonging to the peripheral nervous system (i.e. alpha motorneurones) [140]. This is clinically evident in the assessment of motor function where typically motor weakness of central (increased tone, spasticity, increased reflexes) and peripheral (reduced muscle tone, muscle atrophy, loss of reflexes) origin can be seen in the same patient. In these motor segments (myotomes) originating from areas with cord damage, there is always some alpha motorneurone damage, while below the level of lesion motor weakness is due to the loss of the descending central fibre tracts (i.e. pyramidal spinal tract) [141, 142]. As for the brain, the cord contains neural networks (integration and modulation of in/outputs that affect the facilitation or inhibition of neural inputs) and conductive pathways (longitudinal ascending/ descending fibre tracts). Accordingly, the cord is not only involved in conveying afferent efferent signals but also has a capacity to influence even rather complex sensorimotor functions (like walking) in a subhierarchical capacity relative to the brain. The abovementioned findings indicate that the potential use of stem cells to improve the outcome of human SCI potentially can potentially affect many different aspects of the spinal cord.
So far there is no approved or established treatment of the injured spinal cord itself and all the success achieved to date have been through rehabilitation and improved outcome based on better management of secondary medical problems (like bowel bladder function). Although patients undergoing conventional rehabilitation programmes achieve advanced levels of functional outcome they still have a strong desire to improve their medical condition (patients acknowledge that they learned to live with SCI but they want to go beyond this). Due to these strong emotional desires many patients seek any potential treatment and they may even circumvent regulated (i.e. controlled) health care provisions (many will travel abroad to receive unproven treatments with any kind of cells). They even accept to pay at their own expense, enormous amounts of money (20 000–30 000 USD) to receive these cell applications even though none of the provided interventions have been proven to be effective.
Concepts and preclinical models of cell based therapies in SCI follow in principle the same considerations as in stroke and other CNS disorders (Fig. 14.1). Most commonly applied are preclinical models applying olfactory ensheathing cells (OECs) [143, 144], Schwann cells (SCs) [145, 146], bone marrow stromal cells (BMSCs) [147], and NSCs [148, 149]. While the latter approaches have transferred to a degree to clinical trials, embryonic (ESCs) and induced pluripotent stem cells (iPSCs) although also intensively tested in animal models have not yet reached a required level of safety and confidence for their application in humans outside of a small trial funded by Geron.
The improvement of locomotor recovery and surgical feasibility of cell transplantation has been shown in several experimental paradigms and includes: adult mice neural precursor cells [150]; combined SCs, olfactory ensheathing cells (OEC) [151], and chondroitinase ABC [152]; human SCs [153]: olfactory ensheathing cells [154]; homologous macrophages [155]; human ES oligo progenitors [156]; human umbilical cord cells [157]; human neurons from an embryonal teratocarcinoma cell line [158]; human neural Spinal precursor cells [159]; and human adult neural stem cells described herein [160–163].
So far in preclinical models the three most likely mechanisms for using stem cells on the damaged cord include: (1) denovo remyelination, and (2) neurotrophic effects increasing neural plasticity, and (3) replacement of lost cells [164]. These occur to differing extents and the functional readouts in the animal mainly disclose minimal to moderate effects on locomotion. To enhance treatment effects combinatorial interventions are becoming increasingly tested and hold some promise [165, 166].
While in principle the application of stem cells in animal SCI models appears feasible and reasonably safe many important aspects for translating these application into human treatments are unresolved: i) What is the most reasonable animal model (is there a need for nonhuman primate studies)?; ii) What injury model (contusion versus cut lesions) and extent (completeness of cord damage) of cord injury is most relevant?; iii) What kind of cell line may be superior and are there any reliable dose dependencies on outcomes?, and iv) What is the most sensitive timing after injury (what constitutes acute and how is that established in animals and humans) for cell transplantation [167]?
Furthermore, the estimation of potential effect sizes as observed in the animal models to those involving patients is unclear [168]. While in human studies the stratification of patients has been typically based on actual clinical phenotypes on admittance (level and completeness of lesion, time after injury, and accompanying medical complications) novel stratification approaches are based on advanced prediction models that become targeted to the aimed primary endpoint and therefore improve the enrolment of appropriate patients and increase the statistical power [169–171]. In contrast, animal studies often apply a post hoc analysis with a stratification based on performance and biological markers that eventually allows one to find differences in outcomes. Clinical phase I/II trials concentrate on safety and feasibility (route of application, interactions of cells with host, interference of cell transplantation on recovery profiles, etc.) and so far animal models are of limited predictive value in terms of safety concerns [172]. One of the serious anxieties relates to the induction or increase of neuropathic pain, which is frequently an ongoing challenge for patients following SCI (in about 60–70% of patients) [173, 174]. The induction of pain has not only been reported in animal models [175] but also in a case control series of intrathecal autologous bone marrow transplantations in humans with chronic SCI [176, 177].
All the above mentioned issues need to be carefully considered when thinking about the translation of preclinical findings into a clinical trial [167]. In humans the procedural (surgical) and biological (cell integration, immunogenicity) safety of cell transplantation into the spinal cord has been revealed in three recent completed cell based trials applying intramedullary injections of cells [178–180]. Although the applied cells were of various types of non CNS autografts, the overall findings revealed the general feasibility of the approach and the surgical risk of cell implantation into the injured spinal cord was considered favourable. The first trial was a phase I study performed in Israel and assessed the safety of implanting incubated autologous macrophages within 14 days of injury [179]. The premise for this study was based on the concept of ‘protective autoimmunity’ in which endogenous activated macrophages and T cells are assumed to help augment spinal cord repair in the subacute inflammatory phase (1–2 weeks post injury). The study enrolled eight patients with complete injury between C5 andT11 and the intervention consisted of four microinjections (60 microlitres) of autologous harvested macrophages (total 4 million cells) at the caudal border of the cord injury. No adverse events were attributed to the experimental therapy, and no acute or delayed morbidity associated with the volume or cell dose injected into the spinal cord was observed. This study resulted in a phase II study that enrolled 50 subjects before study cancellation for financial reasons. The safety and efficacy data from this study will be analysed and eventually published [181].
The second cell based study involved implantation of autologous bone marrow cells in combination with systemic granulocyte macrophage colony stimulating factor (GMCSF) administration in a phase I/II trial [180]. In this study a series of 35 patients with complete cervical or thoracic injuries were implanted with autologous bone marrow cells at various stages after injury (acute, subacute, and chronic). The premise of the study was based on the possibility of bone marrow derived cells producing neuroprotective cytokines or differentiating into neural cells helpful for repair. The surgical procedure involved exposing the injured cord and injecting 200 million cells in a total suspension volume of 1.8 ml (six 300 microlitre aliquots) ‘surrounding the lesion site’. One patient reported a transient postoperative reduction in hand strength and three patients had increased incisional muscle rigidity (presumably related to the surgical exposure). The authors reported that neuropathic pain was observed in a higher proportion (20%) of the patients who underwent transplantation, as opposed to the parallel ‘control’ (nontransplanted) patient group (7.7%). The increased neuropathic pain was predominantly noted in those patients transplanted in the subacute and chronic stage. The quality and nature of the neuropathic pain was not fully characterized in the report and pretransplant pain assessments were not quantified. In addition, the confounding variable of the second surgery necessary for the bone marrow cell transplantation as compared to the single stabilization surgery performed in the control group was not accounted for in the analysis of the neuropathic pain. The third study using a cell based strategy for SCI involved the injection of autologous olfactory ensheathing cells (OES) in three patients with a complete thoracic cord injury [178]. The cell doses in this limited series ranged between 12 and 28 million cells and were injected into the area of injury and adjacent cord using a pattern between 270 and 630 spinal cord injections. No deterioration in function or neuropathic pain was reported for the three subjects in this limited trial.
In 2009, the US Food and Drug Administration (FDA) approved a phase I clinical trial (Geron company) to evaluate the safety of a human ES cell based product candidate, GRNOPC1, in patients with acute thoracic spinal cord injuries. The study was open to patients with a neurologically complete (ASIA Impairment Scale A) traumatic SCI limited to the thoracic region between T3 to T11. The administration of GRNOPC1 was supposed to occur between 7 and 14 days after the injury [182]. In total five patients were enrolled in this study until in November 2011 the company, due to strategic considerations, abandoned the study. There have been no serious adverse events reported so far. Meanwhile the company GeronR came insolvent and was replaced by AsteriasR be performing a dose escalation study in cervical sensorimotor complete SCI (Asterias ASTOPC1 started 2015, NCT02302157) [183].
The safety and feasibility of intramedullary transplantation of human NSCs in thoracic (phase I/II thoracic (n = 12), regulated by Swiss medic/Health Canada NCT01321333 started 2011 and completed 2015) and cervical (phase II cervical (n = 17), regulated by FDA NCT02163876) started in 2014 and prematurely terminated in June 2016) SCI has been proven in two clinical studies (sponsored by Stem cells Inc) [184, 185]. Unfortunately, again this company went into liquidation leaving the field without longterm followup studies and pointing to the challenges of advanced clinical trials by middle sized spin off companies [186].
Furthermore, a clinical trial (initiated by The Miami Project to Cure Paralysis NCT01739023 started 2012, completed 2016) with the transplantation of autologous human Schwann cells in subacute thoracic SCI has again shown the surgical feasibility of this approach without efficacy. [187].
The aforementioned findings (the list of studies is by no means considered to be complete) reveal that although there have been a limited number of studies in humans, the application of stem cells for human SCI appears to be feasible. So far, however, there have been no findings in humans that reveal major or clinical immediately obvious improvements in motor or sensory function.
Conclusions
The current possibilities for the structural and functional repair of the injured and diseased CNS are still extremely limited and lesions to the adult brain or spinal cord often result in a detrimental and disabling failure of CNS function. Thus, novel therapeutic avenues are needed. The identification of somatic stem cells within the adult nervous tissue and the improved handling and generation of various multipotent and pluripotent human stem cells, has raised hopes that these cells, whether endogenous or transplanted, will be useful for tissue repair. Even though stem cells are today not routinely used in the clinics to treat CNS diseases, a growing number of clinical studies have identified their great potential for functional repair or support of injured tissue. Ongoing studies of stem cell based treatments at this time are starting to explore their tolerability and to some extent efficacy. It seems plausible that diseases with a rather defined pathology (i.e. the loss of distinct cell populations such as dopaminergic cells in the substantia nigra in PD) represent more promising targets compared to those with more diffuse neural tissue damage occurring for example after stroke. Further, disease stratification, based on the identification of patient subgroups that may benefit more than others, will be important to ultimately judge the potential of stem cell therapies for several neuropsychiatric diseases. Clearly, future preclinical and clinical studies will have to identify the appropriate source for transplanted cells, be they patient derived such as iPSCs or derived from other human tissue representing allografts. Safety, comparability, and largescale availability of cells are certainly a prerequisite for the routine clinical use of stem cells or their derivatives. Important for the field and subsequently the clinical success of stem cell based therapies will be the need to avoid premature and over optimistic expectations of their efficacy as we move towards patients in the clinic.
REFERENCES
1. Spalding KL, Bergmann O, Alkass K, Bernard S, et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153(6):1219–92.
2. Barker RA, Gotz M, Parmar M. New approaches for brain repair-from rescue to reprogramming. Nature. 2018;557:329–34.
3. Barker RA, Barrett J, Mason SL, Bjorklund A. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s dis-ease. J Neurol Neurosurg Psychiatry. 2013;84(6):657–65.
4. Barker RA, Drouin-Ouellet J, Parmar M. Cell-based therapies for Parkinson disease-past insights and future potential. Nat Rev Neurol. 2015;11:492–503.
5. Altman J, Das GD. Post-natal origin of microneurons in the rat brain. Nature. 1965;207:953–6.
6. Kaplan MS, Hinds JW. Neurogenesis in the adult rat: elec-tron microscopic analysis of light radioautographs. Science. 1977;197:1092–94.
7. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–10.
8. Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–33.
9. Gage F. Mammalian neural stem cells. Science. 2000;287:1433–8.
10. Zhao C, Deng W, Gage FH. Mechanisms and functional implica-tions of adult neurogenesis. Cell. 2008;132:645–60.
11. Pilz GA, Bottes S, Betizeau M, et al. Live imaging of neurogenesis in the adult mouse hippocampus. Science. 2018;359:658–62.
12. Armstrong RJ, Barker RA. Neurodegeneration: a failure of neuroregeneration? Lancet. 2011;358:1174–6.
13. Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nature Neurosci. 2007;10:1110–15. 14. Kriks S, Shim JW, Piao J, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–51.
15. Kirkeby A, Grealish S, Wolf DA, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Reports.
2012;1:703–14.
16. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripo-tent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
17. Cicchetti F, Soulet D, Freeman TB. Neuronal degeneration in striatal transplants and Huntington’s disease: potential mechan-isms and clinical implications. Brain. 2011;134(Pt 3):641–52.
18. Gould E, Tanapat P. Stress and hippocampal neurogenesis. Biol Psychiatry. 1999;46:1472–9.
19. Anacker C, Hen R. Adult hippocampal neurogenesis and cogni-tive flexibility—linking memory and mood. Nat Rev Neurosci. 2017;18:335–46.
20. Clark RE, Broadbent NJ, Zola SM, Squire LR. Anterograde am-nesia and temporally graded retrograde amnesia for a nonspatial memory task after lesions of hippocampus and subiculum. J Neurosci.
2002;22:4663–9.
21. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–9.
22. Sahay A, Scobie KN, Hill AS, et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–70
23. Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychology. 1979;93:74–104.
24. Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40.
25. Sorrells SF, Paredes MF, Cebrian-Silla A, et al. Human hippocampal neurogenesis drops sharply in children to un-detectable levels in adults. Nature. 2018;555(7696):377–81.
26. Boldrini M, Fulmore CA, Tartt AN, et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell. 2018;22:589–99.e5.
27. Kempermann G, Gage FH, Aigner L, et al. Human adult neuro-genesis: evidence and remaining questions. Cell Stem Cell. 2018;23(1):25–30.
28. Drapeau E, Mayo W, Aurousseau C, Le Moal M, Piazza PV, Abrous DN. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2003;100:14385–90.
29. Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12:585–601.
30. Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogen-esis by long-term environmental enrichment. Ann Neurol. 2002;52:135–43.
31. van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–5.
32. Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38.
33. Jessberger S, Zhao C, Toni N, Clemenson GD, Jr., Li Y, Gage FH. Seizure-associated, aberrant neurogenesis in adult rats char-acterized with retrovirus-mediated cell labeling. J Neurosci.
2007;27:9400–7.
34. Walter C, Murphy BL, Pun RY, Spieles-Engemann AL, Danzer SC. Pilocarpine-induced seizures cause selective time-dependent changes to adult-generated hippocampal dentate granule cells. J Neurosci.
2007;27:7541–52.
35. Pun RY, Rolle IJ, Lasarge CL, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epi-lepsy. Neuron. 2012;75:1022–34.
36. Scharfman HE, Hen R. Neuroscience. Is more neurogenesis al-ways better? Science. 2007;315:336–8. 37. Thodeson DM, Brulet R, Hsieh J. Neural stem cells and epi-lepsy: functional roles and disease-in-adish models. Cell Tissue Res. 2018;371:47–54.
38. Shetty AK. Progress in cell grafting therapy for temporal lobe epilepsy. Neurotherapeutics. 2011;8:721–35.
39. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol. 2002;249 Suppl 3:III/ 1–5.
40. Chaudhuri KR, Odin P. The challenge of non-motor symptoms in Parkinson’s disease. Prog Brain Res. 2010;184:325–41.
41. Mahlknecht P, Gasperi A, Willeit P, et al. Prodromal Parkinson’s disease as defined per MDS research criteria in the general eld-erly community. Mov Disord. 2016;31:1405–8.
42. Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci.
2017;18:509
43. Brundin P, Kordower JH. Neuropathology in transplants in Parkinson’s disease: implications for disease pathogenesis and the future of cell therapy. Prog Brain Res. 2012;200:221–41.
44. Brundin P, Melki R. Prying into the prion hypothesis for Parkinson’s disease. J Neurosci. 2017;37:9808–18.
45. Surmeier DJ, Obeso JA, Halliday GM. Parkinson’s disease Is not simply a prion disorder. J Neurosci. 2017;37:9799–807.
46. Lubbe S, Morris HR. Recent advances in Parkinson’s disease gen-etics. J Neurol. 2014;261(2):259–66. 47. Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 2013;136:392–9.
48. Liu G, Locascio JJ, Corvol JC, et al. Prediction of cognition in Parkinson’s disease with a clinical-genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol. 2017;16:620–9.
49. Chang D, Nalls MA, Hallgrimsdottir IB, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49:1511–16.
50. Williams-Gray CH, Evans JR, Goris A, et al. The distinct cog-nitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132:2958–69.
51. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and de-mentia in patients with Parkinson’s disease. Lancet Neurol. 2010;9:1200–13.
52. Drouin-Ouellet J, Barker RA. Parkinson’s disease in a dish: what patient specific-reprogrammed somatic cells can tell us about Parkinson’s disease, if anything? Stem Cells Int. 2012:926147.
53. Dyson SC, Barker RA. Cell-based therapies for Parkinson’s dis-ease. Expert Rev Neurother. 2011;11:831–44.
54. Daley GQ. The promise and perils of stem cell therapeutics. Cell Stem Cell. 2012;10:740–9. 55. Mertens J, Paquola ACM, Ku M, et al. Directly reprogrammed human neurons retain agingassociated transcriptomic signa-tures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell.
2015;17:705–18.
56. Prakash N, Wurst W. Development of dopaminergic neurons in the mammalian brain. Cell Mol Life Sci. 2006;63:187–206.
57. Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(Pt 8):1437–48.
58. Soldner F, Hockemeyer D, Beard C, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral repro-gramming factors. Cell. 2009;136:964–77.
59. Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–95.
60. Kouroupi G, Taoufik E, Vlachos IS, et al. Defective synaptic con-nectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc Natl Acad Sci U S A. 2017;114:E3679– 88.
61. Chung SY, Kishinevsky S, Mazzulli JR, et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mito-chondrial dysfunction and alpha-synuclein accumulation. Stem Cell Reports.
2016;7:664–77.
62. Hsieh CH, Shaltouki A, Gonzalez AE, et al. Functional impair-ment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016;19:709–24.
63. Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum
Mol Genet. 2009;18:R48–59
64. Miura K, Okada Y, Aoi T, et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol. 2009;27:743–5.
65. Barker RA, Parmar M, Studer L, Takahashi J. Human trials of stem cell-derived dopamine neurons for Parkinson’s dis-ease: dawn of a new era. Cell Stem Cell. 2017;21:569–73.
66. Kikuchi T, Morizane A, Doi D, et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature. 2017;548:592–6.
67. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–41.
68. Pfisterer U, Kirkeby A, Torper O, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A. 2011;108:10343–8.
69. Qiang L, Fujita R, Yamashita T, et al. Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell. 2011;146(3):359–71.
70. Caiazzo M, Dell’Anno MT, Dvoretskova E, et al. Direct gen-eration of functional dopaminergic neurons from mouse and human fibroblasts. Nature. 2011;476:224–7.
71. Kim J, Su SC, Wang H, et al. Functional integration of dopamin-ergic neurons directly converted from mouse fibroblasts. Cell Stem Cell. 2011;9:413–19.
72. Liu X, Li F, Stubblefield EA, et al. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res. 2012;22:321–32.
73. Shrigley S, Pircs K, Barker RA, Parmar M, Drouin-Ouellet J. Simple generation of a high yield culture of induced neurons from human adult skin fibroblasts. J Vis Exp. 2018;(132). doi: 10.3791/56904.
74. Brundin P, Barker RA, Parmar M. Neural grafting in Parkinson’s disease Problems and possibilities. Prog Brain Res. 2010;184:265–94.
75. Kordower JH, Rosenstein JM, Collier TJ, et al. Functional fetal nigral grafts in a patient with Parkinson’s dis-ease: chemoanatomic, ultrastructural, and metabolic studies. J Comp Neurol.
1996;370:203–30.
76. Galpern WR, Corrigan-Curay J, Lang AE, et al. Sham neuro-surgical procedures in clinical trials for neurodegenerative diseases: scientific and ethical considerations. Lancet Neurol. 2012;11:643–50.
77. Freed CR, Greene PE, Breeze RE, et al. Transplantation of embry-onic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710–19.
78. Olanow CW, Goetz CG, Kordower JH, et al. A double-blind con-trolled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403–14.
79. Olanow CW, Kordower JH, Lang AE, Obeso JA. Dopaminergic transplantation for Parkinson’s disease: current status and future prospects. Ann Neurol. 2009;66:591–6.
80. Barker RA, Barrett J, Mason SL, Bjorklund A. Fetal dopamin-ergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013;12:84–91.
81. Barker RA, de Beaufort I. Scientific and ethical issues related to stem cell research and interventions in neurodegenerative dis-orders of the brain. Prog Neurobiol. 2013;110;63–73.
82. Lerou PH, Daley GQ. Therapeutic potential of embryonic stem cells. Blood Rev. 2005;19:321–31. 83. Kirkeby A, Nolbrant S, Tiklova K, et al. Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-based therapy for Parkinson’s disease. Cell Stem Cell.
2017;20:135–48
84. Cai J, Yang M, Poremsky E, Kidd S, Schneider JS, Iacovitti L. Dopaminergic neurons derived from human induced pluripo-tent stem cells survive and integrate into 6-OHDA-lesioned rats. Stem Cells Dev.
2010;19:1017–23.
85. Wernig M, Zhao JP, Pruszak J, et al. Neurons derived from re-programmed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:5856–61.
86. Rivetti di Val Cervo P, Romanov RA, Spigolon G, et al. Induction of functional dopamine neurons from human astro-cytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat Biotechnol.
2017;35:444–52.
87. Phillips W, Shannon KM, Barker RA. The current clinical man-agement of Huntington’s disease. Mov Disord. 2008;23:1491–504.
88. Ross CA, Tabrizi SJ. Huntington’s disease: from mo-lecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98.
89. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12(7):637–49.
90. Ernst A, Alkass K, Bernard S, et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–83.
91. Clelland CD, Barker RA, Watts C. Cell therapy in Huntington disease. Neurosurg Focus. 2008;24:E9. 92. Dunnett SB, Carter RJ, Watts C, et al. Striatal transplantation in a transgenic mouse model of Huntington’s disease. Exp Neurol. 1998;154:31–40.
93. Cisbani G, St. Pierre M, Cicchetti F. Single cell suspension methodology favours survival and vascularization of fetal stri-atal grafts in the YAC128 mouse model of Huntington’s disease. Cell Transplant. 2014;23(10):1267–78.
94. Wijeyekoon R, Barker RA. The current status of neural grafting in the treatment of Huntington’s disease. A review. Front Integr Neurosci. 2011;5:78.
95. Bachoud-Levi AC, Deglon N, Nguyen JP, et al. Neuroprotective gene therapy for Huntington’s disease using a polymer encap-sulated BHK cell line engineered to secrete human CNTF. Hum Gene Ther.
2000;11:1723–9.
96. Hauser RA, Sandberg PR, Freeman TB, Stoessl AJ. Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology. 2002;58:1704; author reply 1704.
97. Cisbani G, Cicchetti F. The fate of cell grafts for the treatment of Huntington’s disease: the postmortem evidence. Neuropathol Appl Neurobiol. 2014;40:71–90.
98. Reuter I, Tai YF, Pavese N, et al. Long-term clinical and posi-tron emission tomography outcome of fetal striatal transplant-ation in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2008;79:948–51. 99. Li M, Rosser AE. Pluripotent stem cell-derived neurons for transplantation in Huntington’s disease. Prog Brain Res. 2017;230:263–81.
100. Ma L, Hu B, Liu Y, et al. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–64.
101. Carri AD, Onorati M, Lelos MJ, et al. Developmentally coord-inated extrinsic signals drive human pluripotent stem cell dif-ferentiation toward authentic DARPP-32+ medium-sized spiny neurons.
Development. 2013;140:301–12.
102. Arber C, Precious SV, Cambray S, et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–86
103. Reidling JC, Relaño-Ginés A, Holley SM, et al. Human neural stem cell transplantation rescues functional deficits in R6/2 and Q140 Huntington’s disease mice. Stem Cell Rep. 2018;10:58–72. 104. Benraiss A, Wang S, Herrlinger S, et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun. 2016;7:11758.
105. The HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–78.
106. Phillips W, Morton AJ, Barker RA. Abnormalities of neuro-genesis in the R6/2 mouse model of Huntington’s disease are attributable to the in vivo microenvironment. J Neurosci. 2005;25:11564–76. 107. Clelland CD, Choi M, Romberg C, et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–13.
108. Begeti F, Schwab LC, Mason SL, Barker RA. Hippocampal dys-function defines disease onset in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2016;87:975–81.
109. Curtis MA, Penney EB, Pearson AG, et al. Increased cell prolif-eration and neurogenesis in the adult human Huntington’s dis-ease brain. Proc Natl Acad Sci U S A. 2003;100:9023–7.
110. O’Keeffe GC, Tyers P, Aarsland D, Dalley JW, Barker RA, Caldwell MA. Dopamine-induced proliferation of adult neural precursor cells in the mammalian subventricular zone is medi-ated through EGF. Proc Natl Acad Sci U S A. 2009;106:8754–9.
111. Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;5:5.
112. Jin K, Wang X, Xie L, et al. Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A. 2006;103:13198–202.
113. Lindvall O, Kokaia Z. Stem cells in human neurodegenerative disorders—time for clinical translation? J Clin Invest. 2006;120:29–40.
114. Kalladka D, Sinden J, Pollock K, et al. Human neural stem cells in patients with chronic ischaemic stroke (PISCES): a phase 1, first-in-man study. Lancet. 2016;388:787–96.
115. Bhasin A, Srivastava MV, Kumaran SS, et al. Autologous mes-enchymal stem cells in chronic stroke. Cerebrovasc Dis Extra. 2011;1:93–104.
116. Connick P, Kolappan M, Crawley C, et al. Autologous mesen-chymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol.
2012;11(2):150–6.
117. Rice CM, Mallam EA, Whone AL, et al. Safety and feasibility of autologous bone marrow cellular therapy in relapsing-progressive multiple sclerosis. Clin Pharmacol Ther. 2010;87(6):679–85.
118. Scolding NJ, Pasquini M, Reingold SC, et al. Cell-based thera-peutic strategies for multiple sclerosis.
Brain. 2017;140:2776–96.
119. Martino G, Pluchino S. The therapeutic potential of neural stem cells. Nat Rev Neurosci.
2006;7:395–406.
120. Jadasz JJ, Aigner L, Rivera FJ, Kury P. The remyelination Philosopher’s Stone: stem and progenitor cell therapies for mul-tiple sclerosis. Cell Tissue Res. 2012;349:331–47.
121. Rafalski VA, Ho PP, Brett JO, et al. Expansion of oligodendro-cyte progenitor cells following SIRT1 inactivation in the adult brain. Nat Cell Biol. 2013;15(6):614–24
122. Huang JK, Fancy SP, Zhao C, Rowitch DH, French-Constant C, Franklin RJ. Myelin regeneration in multiple sclerosis: targeting endogenous stem cells. Neurotherapeutics. 2011;8:650–8. 123. Murphy NA, Franklin RJM. Recruitment of endogenous CNS stem cells for regeneration in demyelinating disease. Prog Brain Res. 2017;231:135–63.
124. Lee PH, Lee JE, Kim HS, et al. A randomized trial of mesen-chymal stem cells in multiple system atrophy. Ann Neurol. 2012;72:32–40.
125. Mazzini L, Vercelli A, Ferrero I, Boido M, Cantello R, Fagioli F. Transplantation of mesenchymal stem cells in ALS. Prog Brain Res. 2012;201:333–59.
126. Cripps RA, Lee BB, Wing P, Weerts E, Mackay J, Brown D. A global map for traumatic spinal cord injury epidemiology: to-wards a living data repository for injury prevention. Spinal Cord.
2011;49(4):493–501.
127. Lee BB. The global map for traumatic spinal cord injury epi-demiology: update 2011, global incidence rate. Spinal Cord. 2014;52(2):110–16.
128. Middleton JW, Hontecillas R, Horne WT, et al. Life expect-ancy after spinal cord injury: a 50-year study. Spinal Cord. 2012;250(11):803–11.
129. Ahuja CS, Wilson JR, Nori S, et al. Traumatic spinal cord injury. Nat Rev Dis Primers. 2017;3:17018. 130. Meinecke FW, Exner G. Treatment of patients with spinal cord lesions in Germany 1996-state of the art. Spinal Cord. 1997;35(7):411.
131. Schultke E, Guttmann L. Emerging concept of rehabilitation after spinal cord injury. J Hist Neurosci. 2001;10(3):300–7.
132. Furlan JC, Hitzig SL, Craven BD. The influence of age on func-tional recovery of adults with spinal cord injury or disease after inpatient rehabilitative care: a pilot study. Aging Clin Exp Res.
2013;25(4):463–71.
133. Shin JC, Kim DH, Yu SJ, Yang HE, Yoon SY. Epidemiologic change of patients with spinal cord injury.
Ann Rehabil Med. 2013;37(1):50–6.
134. Wirz M, Dietz V. Concepts of aging with paralysis: impli-cations for recovery and treatment. Handb Clin Neurol. 2012;109:77–84.
135. Freund P, Weiskopf N, Ward NS, et al. Disability, atrophy and cortical reorganization following spinal cord injury. Brain. 2011;134:1610–22.
136. Freund P, Weiskopf N, Ashburner J, et al. MRI investigation of the sensorimotor cortex and the corticospinal tract after acute spinal cord injury: a prospective longitudinal study. Lancet Neurol.
2013;12:873–81.
137. Grabher P, Callaghan MF, Ashburner J, et al. Tracking sensory system atrophy and outcome prediction in spinal cord injury. Ann Neurol. 2015;78:751–61.
138. Grabher P, Blaiotta C, Ashburner J, Freund P. Relationship between brainstem neurodegeneration and clinical impair-ment in traumatic spinal cord injury. Neuroimage Clin. 2017;15:494–501. 139. Ziegler G, Grabher P, Thompson A, et al. Progressive neurodegeneration following spinal cord injury: implications for clinical trials. Neurology. 2018;90:e1257–66.
140. Freund P, Friston K, Thompson AJ, et al. Embodied neur-ology: an integrative framework for neurological disorders. Brain. 2016;139:1855–61.
141. Dietz V, Curt A. Neurological aspects of spinal-cord re-pair: promises and challenges. Lancet Neurol.
2006;5(8):688–94
142. Petersen JA, Spiess M, Curt A, et al. Upper limb recovery in spinal cord injury: involvement of central and peripheral motor pathways. Neurorehabil Neural Repair. 2017;31:432–41.
143. Kalincik T, Jozefcikova K, Sutharsan R, Mackay-Sim A, Carrive P, Waite PM. Selected changes in spinal cord morphology after T4 transection and olfactory ensheathing cell transplantation. Auton Neurosci. 2010;158(1–2):31–8.
144. Zhang J, Wang B, Xiao Z, et al. Olfactory ensheathing cells promote proliferation and inhibit neuronal differentiation of neural progenitor cells through activation of Notch signaling. Neuroscience.
2008;153(2):406–13.
145. Wills TE, Batchelor EP, Kerr NF, et al. Corticospinal tract sprouting in the injured rat spinal cord stimulated by Schwann cell preconditioning of the motor cortex. Neurol Res. 2013;35(7):763–72 146. Guest JD, Hiester ED, Bunge RP. Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Exp Neurol. 2005;192(2):384–93. 147. Alexanian AR, Fehlings MG, Zhang Z, Maiman DJ. Transplanted neurally modified bone marrowderived mesen-chymal stem cells promote tissue protection and locomotor re-covery in spinal cord injured rats. Neurorehabil Neural Repair. 2011;25(9):873–80.
148. Harrop JS, Hashimoto R, Norvell D, et al. Evaluation of clinical experience using cell-based therapies in patients with spinal cord injury: a systematic review. J Neurosurg Spine. 2012;17(1 Suppl):230–46. 149. Kumamaru H, Ohkawa Y, Saiwai H, et al. Direct isolation and RNA-seq reveal environmentdependent properties of engrafted neural stem/progenitor cells. Nat Commun. 2012;3:1140. 150. Eftekharpour ES, Karimi-Abdolrezaee S, Fehlings MG. Current status of experimental cell replacement approaches to spinal cord injury. Neurosurg Focus. 2008;24(3–4):E19.
151. Mackay-Sim A, St John JA. Olfactory ensheathing cells from the nose: clinical application in human spinal cord injuries. Exp Neurol. 2011;229(1):174–80.
152. Fouad K, Pearse DD, Tetzlaff W, Vavrek R. Transplantation and repair: combined cell implantation and chondroitinase delivery prevents deterioration of bladder function in rats with complete spinal cord injury. Spinal Cord. 2009;47(10):727–32.
153. Bunge RP. Schwann cells in central regeneration. Ann N Y Acad Sci. 1991;633:229–33.
154. Ramon-Cueto A, Hontecillas R, Horne WT, et al. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron. 2000;25(2):425–35. 155. Rapalino O, Lazarov-Spiegler O, Agranov E, et al. Implantation of stimulated homologous macrophages results in partial re-covery of paraplegic rats. Nat Med. 1998;4(7):814–21. 156. Keirstead HS, Nistor G, Bernal G, et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci.
2005;25(19):4694–705.
157. Saporta S, Kim JJ, Willing AE, Fu ES, Davis CD, Sanberg PR. Human umbilical cord blood stem cells infusion in spinal cord injury: engraftment and beneficial influence on behavior. J Hematother Stem Cell Res. 2003;12(3):271–8.
158. Saporta S, Makoul AS, Willing A, et al. Functional recovery after complete contusion injury to the spinal cord and transplantation of human neuroteratocarcinoma neurons in rats. J Neurosurg. 2002;97(1 Suppl):63–8.
159. Iwanami A, Kaneko S, Nakamura M, et al. Transplantation of human neural stem cells for spinal cord injury in primates. J Neurosci Res. 2005;80(2):182–90.
160. Cummings BJ, Uchida N, Tamaki SJ, et al. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci U S A. 2005;102(39):14069–74. 161. Salazar DL, Uchida N, Hamers FP, Cummings BJ, Anderson AJ. Human neural stem cells differentiate and promote loco-motor recovery in an early chronic spinal cord injury NOD-scid mouse model. PLoS One. 2010;5(8):e12272
162. Hooshmand MJ, Sontag CJ, Uchida N, Tamaki S, Anderson AJ, Cummings BJ. Analysis of hostmediated repair mechan-isms after human CNS-stem cell transplantation for spinal cord injury: correlation of engraftment with recovery. PLoS One. 2011;4(6):e5871.
163. Cummings BJ, Uchida N, Tamaki SJ, Anderson AJ. Human neural stem cell differentiation following transplantation into spinal cord injured mice: association with recovery of loco-motor function. Neurol Res. 2006;28(5):474–81.
164. Ruff CA, Wilcox JT, Fehlings MG. Cell-based transplantation strategies to promote plasticity following spinal cord injury. Exp Neurol. 2012;235(1):78–90.
165. Lu P, Hontecillas R, Horne WT, et al. Computational modeling-based discovery of novel classes of anti-inflammatory drugs that target lanthionine synthetase C-like protein 2. PLoS One.
2012;7(4):e34643.
166. Karimi-Abdolrezaee S, Schut D, Wang J, Fehlings MG. Chondroitinase and growth factors enhance activation and oligodendrocyte differentiation of endogenous neural precursor cells after spinal cord injury. PLoS One. 2012;7(5):e37589.
167. Curt A. The translational dialogue in spinal cord injury re-search. Spinal Cord. 2012;50(5):352–7.
168. Dietz V, Curt A. Translating preclinical approaches into human application. Handb Clin Neurol.
2012;109:399–409.
169. Tanadini LG, Steeves JD, Hothorn T, et al. Identifying homo-geneous subgroups in neurological disorders: unbiased re-cursive partitioning in cervical complete spinal cord injury. Neurorehabil Neural Repair. 2014;28:507–15.
170. Velstra IM, Bolliger M, Tanadini LG, et al. Prediction and strati-fication of upper limb function and self-care in acute cervical spinal cord injury with the graded redefined assessment of strength, sensibility, and prehension (GRASSP). Neurorehabil Neural Repair. 2014;28:632–42.
171. Tanadini LG, Hothorn T, Jones LA, et al. Toward inclusive trial protocols in heterogeneous neurological disorders: prediction-based stratification of participants with incomplete cer-vical spinal cord injury. Neurorehabil Neural Repair. 2015;29:867–77.
172. Fehlings MG, Vawda R. Cellular treatments for spinal cord injury: the time is right for clinical trials.
Neurotherapeutics. 2011;8(4):704–20.
173. Taylor J, Huelbes S, Albu S, Gomez-Soriano J, Penacoba C, Poole HM. Neuropathic pain intensity, unpleasantness, coping strategies, and psychosocial factors after spinal cord injury: an exploratory longitudinal study during the first year. Pain Med. 2012;13(11):1457–68.
174. Zanca JM, Dijkers MP, Hammond FM, Horn SD. Pain and its impact on inpatient rehabilitation for acute traumatic spinal cord injury: analysis of observational data collected in the SCIRehab study. Arch Phys Med Rehabil. 2013;94(4 Suppl):S137–44.
175. Hofstetter CP, Holmstrom NA, Lilja JA, et al. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci. 2005;8(3):346–53. 176. Kishk NA, Gabr H, Hamdy S, et al. Case control series of intra-thecal autologous bone marrow mesenchymal stem cell therapy for chronic spinal cord injury. Neurorehabil Neural Repair.
2010;24(8):702–8.
177. Curt A. Human neural stem cells in chronic spinal cord injury. Expert Opin Biol Ther.
2012;12(3):271–73.
178. Feron F, Perry C, Cochrane J, et al. Autologous olfactory ensheathing cell transplantation in human spinal cord injury. Brain. 2005;128(Pt 12):2951–60.
179. Knoller N, Auerbach G, Fulga V, et al. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine. 2005;3(3):173– 81.
180. Yoon SH. Shim YS, Park YH, et al. Complete spinal cord injury treatment using autologous bone marrow cell transplantation and bone marrow stimulation with granulocyte macrophage-colony
stimulating factor: phase I/II clinical trial. Stem Cells. 2007;25(8):2066–73
181. Lammertse DP, Jones LA, Charlifue SB, et al. Autologous in-cubated macrophage therapy in acute, complete spinal cord injury: results of the phase 2 randomized controlled multicenter trial. Spinal Cord.
2012;50(9):661–71.
182. Lebkowski J. GRNOPC1: the world’s first embryonic stem cell-derived therapy. Interview with Jane Lebkowski. Regen Med. 2011;6:11–13.
183. Manley NC, Priest CA, Denham J, Wirth ED, 3rd, Lebkowski JS. Human embryonic stem cell-derived oligodendrocyte pro-genitor cells: preclinical efficacy and safety in cervical spinal cord injury. Stem Cells Transl Med. 2017;6:1917–29.
184. Ghobrial GM, Anderson KD, Dididze M, et al. Human neural stem cell transplantation in chronic cervical spinal cord in-jury: functional outcomes at 12 months in a phase II clinical trial. Neurosurgery.
2017;64:87–91.
185. Levi AD, Okonkwo DO, Park P, et al. Emerging safety of intramedullary transplantation of human neural stem cells in chronic cervical and thoracic spinal cord injury. Neurosurgery. 2018;82:562–75. 186. Curt A, Levi AD, Schwab JM. Challenges to translation and the hippocratic oath by premature termination of spinal cord stem cell-based trials. JAMA Neurol. 2017;74:635–6.
187. Anderson KD, Guest JD, Dietrich WD, et al. Safety of autolo-gous human schwann cell transplantation in subacute thoracic spinal cord injury. J Neurotrauma. 2017;34:2950–63